
Band gap and Schottky barrier engineered photocatalyst with promising solar light activity for water remediation
 a
a
Abstract
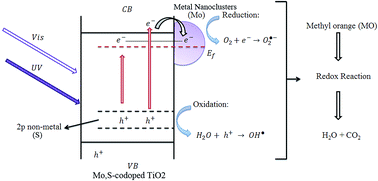
A novel nanophotocatalyst of Mo- and S-doped TiO2 (Mo,S-codoped TiO2) was synthesized via a modified sol–gel method in conjunction with photochemical reduction. The XRD results showed that all of the prepared nanocatalysts only contain the anatase phase. The SEM and TEM analyses revealed that the doping of Mo and S does not cause any change in the morphology of the TiO2 catalyst. Chemical composition analysis carried out using EDX confirmed the successful doping of TiO2 by Mo and S, and DRS illustrated band gap reduction in TiO2 after doping. The photocatalytic performance of the samples was tested using the degradation of methyl orange (MO) as a model organic pollutant. The results showed that the photocatalytic activity of the Mo(0.06%), S(0.05%)-codoped TiO2 catalyst was higher than that of other catalysts under UV and visible light irradiation. Indeed, due to the synergetic effect of doping S and Mo, the Mo,S-codoped TiO2 catalyst has a higher photoactivity than the pure TiO2 and TiO2 doped solely with either Mo or S. Finally, it is proposed that the newly developed Mo,S-codoped TiO2 photocatalyst could be a great candidate for environmental applications such as air and water purification.
 Please wait while we load your content...
Please wait while we load your content...

