
Chemical substitution assisted ion sensing with organic molecules: a case study of naphthalene
Wei-Jie Mina,
Hua Haoa,
Xian-Long Wanga,
Xiao-Hong Zheng*ab and
Zhi Zengab
aKey Laboratory of Materials Physics, Institute of Solid State Physics, Chinese Academy of Sciences, Hefei 230031, China. E-mail: xhzheng@theory.issp.ac.cn; Fax: +86-551-65591434; Tel: +86-551-65591150
bUniversity of Science and Technology of China, Hefei 230026, China
First published on 21st December 2015
Abstract
The chemical modification effects on the electron transport of organic molecules are investigated using first-principles calculations combined with a non-equilibrium Green’s function technique, taking the naphthalene (C10H8) molecule as an example. Particularly, one of the –CH groups in each of the benzene rings is replaced by a N atom. It is found that N substitution greatly increases the quantum conductance by ∼80% due to the reduced HOMO (Highest Occupied Molecular Orbital)–LUMO (Lowest Unoccupied Molecular Orbital) gap and the increased charge transfer from the leads to the molecule. More interestingly, the adsorption of different monovalent cations (H+, Li+, Na+, and K+) to the chemically active N site induces a very different electrical response. To be specific, Li+, Na+, and K+ adsorptions result in a very good conducting state, while H+ adsorption gives rise to an insulating state, providing a promising method for H+ sensing or detection.
1 Introduction
Electron transport in nanoscale and molecular devices has attracted intensive research interest for quite a few decades,1–8 not only because such devices may act as potential candidates for the substitution of traditional silicon based electronic elements so that the lifespan of Moore’s law can be extended in modern electronics, but also because they provide a very good platform for probing the physical laws and nature at the molecular level. As a matter of fact, many interesting transport behaviors have been observed in such devices, such as rectification,9,10 negative differential resistance,11,12 spin filtering,13–15 etc., which arise from the special electronic structures of the various systems and their responses under specific external conditions. More important is that the electron transport can be modulated, fine tuned or even designed purposively in numerous ways, such as via doping, chemical modification, molecular adsorption, electrical and magnetic fields, etc.16–18 Even the manipulation of single or a few atoms in a nanoscale or molecular system may bring about drastic changes and very often completely different transport behaviors can be achieved. We can list with great ease a lot of such typical cases. For example, the substitution of two carbon atoms by a boron (B) atom and a nitrogen (N) atom combined with a transverse electrical field results in electrical switching in metallic carbon nanotubes.17 In zigzag-edged graphene nanoribbons, such a substitution scheme of B and N atoms for a small number of C atoms may lead to 100% spin filtering.16 Doping differently the two C60 molecules of a C60 bridge with B and N atoms can even bring about current rectification.19In recent years, most of the attention in the study of tuning the transport properties at the nanoscale and molecular scale has been focused on the “hotly” studied nanostructures or low dimensional systems, such as fullerenes,20 carbon nanotubes,21 graphene and other 2D systems, like h-BN, MoS2, black phospherene, etc.,22–25 while much less attention has been paid to the tuning of organic molecules, which were the main objects of interest in the earlier period after molecular electronics were proposed. In fact, the first model molecular rectifier proposed by Aviram and Ratner was of an “A-Σ-D” type organic molecule.8 Many prototypical devices showing the basic functions of electronic elements, experimentally or theoretically, are based on organic molecules. Although a lot of studies have been performed with organic molecules, most of them focus on discussing the conductivity of the molecules as a whole and discovering “new” molecules that can realize specific functions. Obviously, the tuning of the transport properties of organic molecules and extending their application are very important in molecular electronics.
In this work, by taking a naphthalene (C10H8) molecule sandwiched between two aluminum electrodes as an example, we will show how the electron transport of organic molecules can be tuned by chemical substitution. Particularly, one of the –CH groups from each of the benzene rings is replaced by a N atom. If a –CH group from only one benzene ring of the molecule is substituted by a N atom, we get a molecule called quinoline. We believe that, with the development of experimental techniques, substitution of the –CH groups in both rings by N atoms can also be achieved. Further, we will study how the electron transport in this system will respond to the adsorption of monovalent ions such as H+, Li+, Na+, and K+ to the chemically active N site. It is found that N substitution increases the equilibrium conductance by a factor of 80% as a result of the reduced HOMO (Highest Occupied Molecular Orbital)–LUMO (Lowest Unoccupied Molecular Orbital) gap and the increased charge transfer from the electrodes to the central molecule. Due to the introduction of the active N site, the adsorption of other ions becomes possible and we find that H+ nearly completely blocks the transmission, which is very different to other studied ions which all keep the system in a good conducting state.
The rest of this paper is organized as follows: in Section II, we give a brief description of the geometry model and of the computational method, while in Section III the main results are presented and discussed. Finally, in Section IV we draw our conclusions.
2 Computational details
The device model consists of a naphthalene molecule sandwiched between two aluminum nanowires, acting as electrodes, through a sulfur atom on each side (see Fig. 1). In each benzene ring, we consider one –CH group being substituted by a N atom, and further, a monovalent atom (H+, Li+, Na+, and K+) is adsorbed onto the N site. Before being connected to the electrodes, the central molecule was fully relaxed by density functional theory (DFT) using the projector augmented plane wave (PAW) method26 as implemented in the VASP package.27 In the geometry relaxations, the plane wave energy cutoff is set to 520 eV, the energy convergence criterion is set to 1.0 × 10−3 and the force criterion is set to 0.001 eV Å−1. | ||
| Fig. 1 The model structure of the device: a naphthalene molecule is sandwiched between two Al(100) electrodes with finite cross sections. The central region includes four and three buffer layers of the left and right electrodes on each side of the molecule, respectively. The colors of the atoms: brown = C, lavender = H, light blue = N, yellow = S; red circles show the positions of the adsorbed monovalent ions H+, Li+, Na+, or K+. | ||
The electron transport was studied using the Nanodcal program, which combines density functional theory and the nonequilibrium Green’s function (NEGF) technique to study the electron transport in molecular devices.28 Using this method, we are able to solve the electron transport problem in an infinite and open system. The two-probe device model is divided into three parts: the left electrode, the right electrode and the central scattering region which includes the molecule and several buffer layers from both electrodes, and these three parts are solved separately. We first calculate the electronic structures of the two electrodes using conventional DFT calculations with localized basis sets and norm-conserving pseudopotentials. From the lead calculations, we get the Hamiltonian matrix and overlap basis matrix and further the self energy matrix of the electrodes. Then, we start the self-consistent calculations for the central region, where the density matrix is calculated using the non-equilibrium Green’s function and the effects of the electrodes are taken into account by the self energy. After the convergence is achieved, the quantities related to electron transport are calculated. The readers interested in the theoretical basis and technique details of this method are referred to the literature.28–30
In our system, the nanowire electrodes have a bulk aluminum structure and extend along the (100) direction. They consist of alternating 5- and 4-atom layers in the fcc geometry. Four atomic layers were chosen as the electrode supercell and the Brillouin zone of the electrodes was sampled by a 1 × 1 × 40 Monkhorst–Pack grid. The ion–electron interaction is described by norm-conserving pseudopotentials and the local density approximation (LDA) is adopted for the exchange–correlation functional. Double-ζ plus polarization (DZP) was chosen as the basis set for all of the atoms.
The transmission function at energy E and bias V was calculated using the Landauer formula31,32
| T(E, V) = Tr(ΓL(E, V)GR(E, V)ΓRGA(E, V)), | (1) |
The current under finite bias is obtained by integrating the transmission function between the bias window [−V/2, V/2] using the formula
 | (2) |
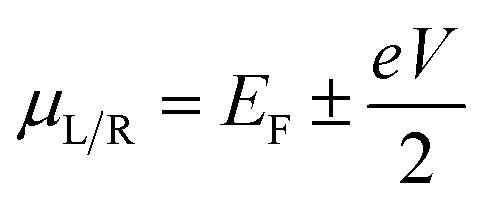 are the chemical potentials and f(E − μL/R) are the Fermi–Dirac distribution functions of the electrons in the two electrodes under non-equilibrium conditions.
are the chemical potentials and f(E − μL/R) are the Fermi–Dirac distribution functions of the electrons in the two electrodes under non-equilibrium conditions.
3 Results and discussion
Since there are many possible configurations when one of the –CH groups is replaced by a N atom in each of the benzene rings, we should figure out which is the one with the lowest energy first. There are 6 inequivalent configurations shown in Fig. 2. These structures have been fully relaxed using the VASP code and their energies are shown in Fig. 3. It is obvious that C-4 has a lower energy than any of the other configurations. We particularly call this configuration C8H6N2. The substitution may result in notable changes to the electronic structure. We are interested in how such changes will be reflected in the electron transmission function. To see this, next, the lowest energy configuration is connected to the two electrodes through S atoms, as shown in Fig. 1. In order to get the most stable molecule–electrode distance, we have calculated the energy of the supercell shown in Fig. 1 as a function of the distance between the surfaces of the two electrodes by full relaxation. The most stable distance is obtained as 11.93 Å. Thus, the following discussions and comparisons of the electron transport are all based on this distance. | ||
| Fig. 2 The six possible configurations when one –CH group is substituted by a N atom in each of the benzene rings of the naphthalene molecule. We name them as C-1, C-2, C-3, …, C-6, respectively, according to their indices in the figure. | ||
 | ||
| Fig. 3 The energy of the six configurations. 1 to 6 represent the corresponding configurations in Fig. 2. The energy of C-4 is taken as a reference by setting it as 0. | ||
The transmission functions of the pristine molecule and the N substituted version are shown in Fig. 4. It is seen that the electron transport in both cases is mediated by the LUMO due to the charge transfer from the electrodes to the molecule, which makes the LUMO partially filled. However, the LUMO transmission peak shifts to a lower energy by 0.35 eV in the N substituted case and thus the equilibrium conductance is increased from 0.17 G0 ( : quantum conductance) to 0.30 G0, a factor of ∼80%. This can be understood by the change to the HOMO–LUMO gap and the different charge transfer. In the pristine C10H8 molecule, the gap is 3.38 eV, while in the C8H6N2 molecule, the gap is 1.82 eV. It is well known that a reduced gap is beneficial for electron transmission. Furthermore, from the Mulliken population analysis, the N substitution increases the charge transfer from 1.1 e to 1.2 e. This also pushes the Fermi level to a higher energy, towards the LUMO. Thus, the transmission is enhanced by the N substitution.
: quantum conductance) to 0.30 G0, a factor of ∼80%. This can be understood by the change to the HOMO–LUMO gap and the different charge transfer. In the pristine C10H8 molecule, the gap is 3.38 eV, while in the C8H6N2 molecule, the gap is 1.82 eV. It is well known that a reduced gap is beneficial for electron transmission. Furthermore, from the Mulliken population analysis, the N substitution increases the charge transfer from 1.1 e to 1.2 e. This also pushes the Fermi level to a higher energy, towards the LUMO. Thus, the transmission is enhanced by the N substitution.
 | ||
| Fig. 4 The transmission functions of the devices with the pristine and the N substituted molecules. The Fermi level is set to 0 throughout the paper. | ||
The impurity atom in a system is often a chemically active site which can easily interact with gas molecules or ions in the environment and thus is intensively studied in many areas like catalysis, hydrogen storage, etc.33–35 For a microscopic system, the adsorption of gas molecules or ions may lead to a characteristic electrical response from the system. This has resulted in new applications for molecular devices in the field of sensors. We now consider the adsorption of monovalent ions (H+, Li+, Na+, and K+) to the N site and see how the transport properties will be affected. Seen in Fig. 5, a common feature in all of these cases is that the LUMO transmission peaks shift to a lower energy. This can be partially understood by the charge transfer shown in Table 1 where we see that more excess electrons are transferred to the organic molecule in the adsorbed cases. Of course, this excess charge has two contributions, one from the electrodes and the other from the adsorbed ion. We also see that due to the repulsion from the electrons transferred from the adsorbed ion, the charge transferred from the electrodes to the molecules greatly decreases. However, the final conductances are quite different. In the Li+, Na+, and K+ cases, all the LUMO peaks are very close to the Fermi level, which results in a high conductance (see Table 1). On the contrary, in the H+ case, the Fermi level is well above and far from the LUMO peak, thus the equilibrium conductance is negligibly small (∼0.01 G0).
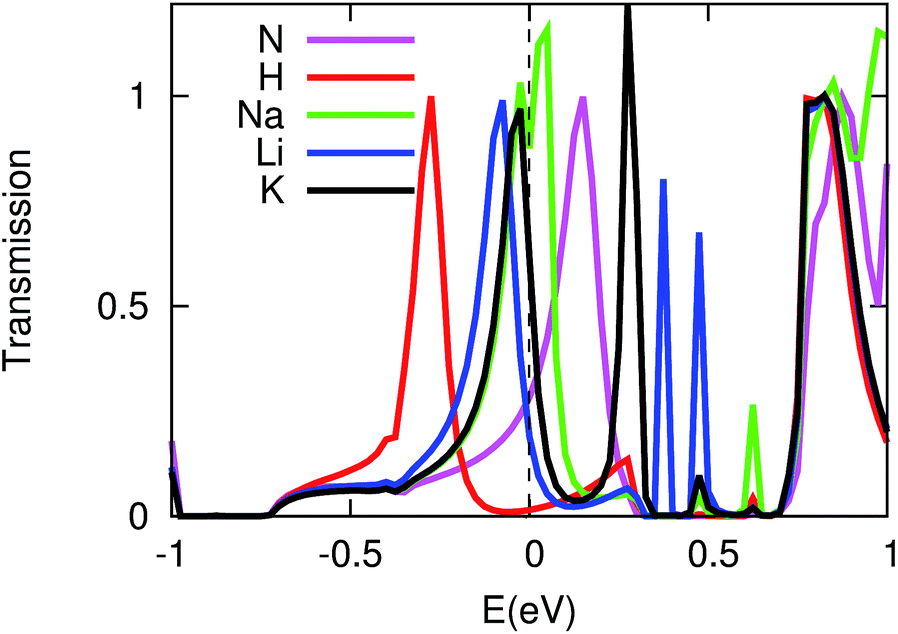 | ||
| Fig. 5 The transmission functions at zero bias. The unadsorbed case and the H+, Na+, Li+, and K+ cases are marked by N, H, Na, Li and K, respectively. | ||
| System | N | Li+ | Na+ | K+ | H+ |
|---|---|---|---|---|---|
| G (G0) | 0.30 | 0.20 | 0.87 | 0.61 | 0.01 |
| ΔQ1 (e) | 1.20 | 0.75 | 0.86 | 0.57 | 0.95 |
| ΔQ2 (e) | — | 1.14 | 0.92 | 1.80 | 0.58 |
| ΔQ (e) | 1.20 | 1.89 | 1.78 | 2.37 | 1.53 |
To gain more insight into the origin of this big difference in the equilibrium conductances, we have studied the localized density of states for all of these cases and they are shown in Fig. 6. From this figure, we can see very intuitively and clearly the positions of the energy levels of the molecules which are indicated by the bright regions in the molecule area. In the H+ case, it is seen that the Fermi level is right in the middle of the LUMO and LUMO+1, which blocks the electron transmission. However, in the other three adsorbed cases, the Fermi level aligns well with the LUMO, thus electrons can easily transmit through the LUMO from one side to the other and consequently a high conductance arises.
 | ||
| Fig. 6 The local density of states (LDOS) at zero bias. (a) Li+, (b) Na+, (c) K+ and (d) H+. For each energy, at each z point, the LDOS is integrated over the xy plane. From this figure, we can see the distribution of the states in both the energy space and the transport direction. | ||
Finally, the different responses of the ion adsorption effects are also reflected in the I–V characteristics, which are shown in Fig. 7. At a low bias (V ≤ 0.5 V), in the unadsorbed case and the Li+, Na+ and K+ cases, the current increases almost linearly with the bias, indicating a very good conducting state. However, in the H+ case, the current is almost zero in the whole bias range studied. Thus, this system can be utilized well for H+ sensing or detection.
 | ||
| Fig. 7 The I–V curves for the N, H+, Na+, Li+, and K+ cases. | ||
4 Conclusion
In summary, we have studied the chemical modification effects on the electron transport of the naphthalene molecule using density functional calculations. It is found that N substitution for a –CH group in each benzene ring can result in the decrease of the HOMO–LUMO gap, which leads to the equilibrium conductance increasing by almost 80%, compared with the pristine molecule. The adsorption of monovalent cations results in different electrical responses. In the H+ case, the system acts as an insulator, while in all of the other cases, a very good conducting state can be observed. Nowadays, besides being candidates for electronic elements in circuits, there is a new trend in the study of molecular electronics for the application of molecular devices in the field of sensing.36–39 For example, the sensing of heavy metal ions in water or soil is a very important step in environmental monitoring and protection. Actually, the usage of molecular devices as sensors for Hg ions and many other heavy metal ions has been well demonstrated experimentally with high efficiency.40 Our study shows a good example of H+ ion sensing which works by using the sharp electrical response induced by the H+ ion adsorption onto the N substituted naphthalene molecule. Although H+ is not a heavy metal atom, this work suggests that besides finding a “new” or “appropriate” molecule for sensing, we should also take into consideration the effect of chemical modification on organic molecules.Acknowledgements
This work was supported by the National Science Foundation of China under Grant No. 11174289, 11374301 and 11574318, Knowledge Innovation Program of Chinese Academy of Sciences, Director Grants of CASHIPS. The calculations were performed in Center for Computational Science of CASHIPS, the ScGrid of Supercomputing Center and Computer Network Information Center of Chinese Academy of Sciences.References
- N. J. Tao, Nat. Nanotechnol., 2006, 1, 173–181 CrossRef CAS PubMed.
- N. Zimbovskaya and M. Pederson, Phys. Rep., 2011, 509, 1–87 CrossRef CAS.
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed.
- S. Karthauser, J. Phys.: Condens. Matter, 2011, 23, 13001–13016 CrossRef PubMed.
- S. Ke, H. Baranger and W. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 085410 CrossRef.
- M. A. Ratner and J. Jortner, Encyclopedia of Physical Science and Technology, 2003, vol. 1991, p. 123139 Search PubMed.
- M. Ratner, Nat. Nanotechnol., 2013, 8, 378–381 CrossRef CAS PubMed.
- A. Aviram and M. A. Ratner, Chem. Phys. Lett., 1974, 29, 277–283 CrossRef CAS.
- D. H. Waldeck and D. N. Beratan, Science, 1993, 261, 576 CAS.
- G. Zhang, M. A. Ratner and M. G. Reuter, J. Phys. Chem. C, 2015, 119, 6254 CAS.
- X. H. Zheng, W. Lu, T. A. Abtew, V. Meunier and J. Bernholc, ACS Nano, 2010, 4, 7205–7210 CrossRef CAS PubMed.
- C. G. Zhang, H. Q. Wang, B. Wang, J. Wang and J. G. Hou, J. Am. Chem. Soc., 2007, 129, 3857–3862 CrossRef PubMed.
- G. Papp and F. M. Peeters, Appl. Phys. Lett., 2001, 78, 2184–2186 CrossRef CAS.
- X. F. Yang, Y. S. Liu, X. Zhang, L. P. Zhou, X. F. Wang, F. Chi and J. F. Feng, Phys. Chem. Chem. Phys., 2014, 16, 11349–11355 RSC.
- P. N. Abufager, R. Robles and N. Lorente, J. Phys. Chem. C, 2015, 119, 12119 CAS.
- X. H. Zheng, R. N. Wang, L. L. Song, Z. X. Dai, X. L. Wang and Z. Zeng, Appl. Phys. Lett., 2009, 95, 123109 CrossRef.
- Y.-W. Son, J. Ihm, M. L. Cohen, S. G. Louie and H. J. Choi, Phys. Rev. Lett., 2005, 95, 216602 CrossRef PubMed.
- X. H. Zheng, J. Lan, X. L. Wang, L. F. Huang, H. Hao and Z. Zeng, Appl. Phys. Lett., 2012, 101, 053101 CrossRef.
- X. H. Zheng, X. L. Wang, Z. X. Dai and Z. Zeng, J. Chem. Phys., 2011, 134, 31–38 Search PubMed.
- V. Alessro, N. D. Treat, J. Jang, C. G. Shuttle, N. A. Batara, F. G. Brunetti, S. J. Hwa, M. L. Chabinyc, C. J. Hawker and A. J. Heeger, Angew. Chem., Int. Ed., 2011, 50, 5166–5169 CrossRef PubMed.
- X. F. Li, K. Q. Chen, L. Wang, M. Q. Long, B. S. Zou and Z. Shuai, Appl. Phys. Lett., 2007, 91, 133511 CrossRef.
- X. H. Zheng, I. Rungger, Z. Zeng and S. Sanvito, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 308–310 Search PubMed.
- Y. Ge, W. Wan, W. Feng, D. Xiao and Y. Yao, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 035414 CrossRef.
- S. Mcdonnell, R. Addou, C. Buie, R. M. Wallace and C. L. Hinkle, ACS Nano, 2014, 8, 2880–2888 CrossRef CAS.
- I. Khan and J. Hong, New J. Phys., 2015, 17, 023056 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- J. Taylor, H. Guo and J. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245407 CrossRef.
- M. Brandbyge, J.-L. Mozos, P. Ordejón, J. Taylor and K. Stokbro, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 165401 CrossRef.
- Y. Xue, S. Datta and M. A. Ratner, Chem. Phys., 2001, 281, 151–170 CrossRef.
- S. Datta, Electronic Transport in Mesoscopic Systems, ed. H. Ahmed, M. Pepper and A. Broers, Cambridge University Press, Cambridge, England, 1995 Search PubMed.
- H. Haug and A.-P. Jauho, Quantum Kinetics in Transport and Optics of Semiconductors, Springer-Verlag, Berlin, 1996 Search PubMed.
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 7698 CrossRef PubMed.
- V. B. Parambhath, R. Nagar and S. Ramaprabhu, Langmuir, 2012, 28, 7826–7833 CrossRef PubMed.
- S. A. Shevlin and Z. X. Guo, Appl. Phys. Lett., 2006, 89, 153104 CrossRef.
- C. J. Chen, S. Manuel and M. A. Ratner, J. Chem. Phys., 2014, 140, 054709 CrossRef PubMed.
- J. X. J. Zhang and K. Hoshino, Molecular Sensors and Nanodevices: Principles, Designs and Applications in Biomedical Engineering, Elsevier, Oxford, 2014 Search PubMed.
- Y. H. Zhang, L. F. Han, Y. H. Xiao, D. Z. Jia, Z. H. Guo and F. Li, Comput. Mater. Sci., 2013, 69, 222–228 CrossRef.
- H. B. Gu, X. Zhang, H. G. Wei, Y. D. Huang, S. Y. Wei and Z. H. Guo, Chem. Soc. Rev., 2013, 42, 5907–5943 RSC.
- Z. Guo, Z. G. Liu, X. Z. Yao, K. S. Zhang, X. Chen, J. H. Liu and X. J. Huang, Sci. Rep., 2013, 3, 1635–1646 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |