
Palladium(ii) and copper(ii) chloride complexes bearing bulky α-diimine ligands as catalysts for norbornene vinyl-addition (co)polymerization†
Abstract
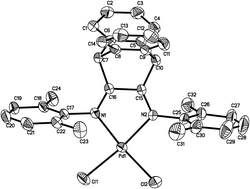
Palladium(II) and copper(II) chloride complexes bearing bulky 9,10-dihydro-9,10-ethanoanthracene-11,12-diimine ligands were synthesized and sufficiently characterized by elemental and spectroscopic analysis along with X-ray diffraction analysis. The X-ray diffraction demonstrated that palladium(II) complexes (C1–C2) and copper(II) complexes (C3–C4) were four-coordinated. All these complexes displayed catalytic activities up to 105 gpolymer molMt−1 h−1 for norbornene vinyl-addition polymerization on treatment with excess methylaluminoxane (MAO). The parameters of the reaction conditions, the type of metals and steric effects of coordinative ligands had influences on the catalytic properties. C2 was selected as a catalyst precursor for the copolymerization of norbornene (NB) with 2-butyloxymethylene norbornene (BN), which exhibited catalytic activities up to 1.1 × 105 gpolymer molPd−1 h−1 and produced copolymers with relatively high molecular weights. The fraction of BN in the copolymer could reach up to 9.8–62.1% by controlling the monomer feed ratio range from 10–80%. The achieved vinyl-addition type poly(NB-co-BN) displayed high thermal stability and was soluble in common organic solvents, such as CHCl3 and THF.
 Please wait while we load your content...
Please wait while we load your content...

