
Low temperature CO catalytic oxidation and kinetic performances of KOH–Hopcalite in the presence of CO2†
Yafei Guoa,
Changhai Lia,
Shouxiang Lu*a and
Chuanwen Zhao*b
aState Key Laboratory of Fire Science, University of Science and Technology of China, Hefei 230026, China. E-mail: sxlu@ustc.edu.cn; Fax: +86 551 63601669; Tel: +86 551 63603141
bJiangsu Provincial Key Laboratory of Materials Cycling and Pollution Control, School of Energy and Mechanical Engineering, Nanjing Normal University, Nanjing 210042, China. E-mail: cwzhao@ustc.edu.cn; Fax: +86 551 63601669; Tel: +86 551 63603141
First published on 13th January 2016
Abstract
Catalytic removal of CO from fire smoke is critical to ensure human safety and post-fire atmospheric recovery in typical confined spaces. Copper manganese oxide compounds show promise as highly efficient catalysts for low temperature CO oxidation. However, the CO oxidation activity will be affected when the catalyst is applied in fire smoke containing high-concentration CO2. In this work, a bi-functional catalyst of KOH–Hopcalite is synthesized by impregnation of KOH on Hopcalite (copper manganese oxides mixture) precursor. The catalyst is characterized by N2 adsorption–desorption, X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FT-IR). CO oxidation activity and long-term working stability of the precursor and catalyst in the presence of CO2 are investigated. CO oxidation activity of the precursor would decrease when CO2 is present. KOH modification can mitigate the inhibiting effect of CO2 on CO oxidation activity of the precursor. Reaction mechanisms and kinetic performances of the catalyst in the presence of CO2 are also demonstrated. The catalyst could be potentially utilized as a scavenging agent for post-fire cleanup and atmospheric recovery in confined spaces.
1. Introduction
Low temperature CO oxidation has received considerable attention due to the potential applications in various fields including automobile exhaust emission control,1 close-cycle CO2 lasers,2 CO gas sensors,3,4 preferential oxidation (PROX) in hydrogen-rich streams,5,6 respiratory protection and indoor air purification.7,8 Recently, the technology option has found a new application for removing trace CO from confined spaces including coal mine refuge chambers, space-crafts and submarines.9–11There are extensive reports highlighting numerous life fatalities in fire accidents.12–14 Particularly, poisoning caused by CO in fire smoke has been considered as the prime reason for life casualties. In typical confined spaces, technology options for smoke management such as natural ventilation or mechanical ventilation are not readily available. In the event of a fire, CO concentration in these spaces will be accumulated to a high level. It is necessary to develop new technology options to remove CO from fire smoke in these spaces to ensure human safety and rapid atmospheric recovery.
Catalytic oxidation into CO2 has been considered as a promising means to CO abatement.15,16 Reports on CO oxidation in confined spaces for post-fire cleanup application are available but limited. NASA evaluated the oxidation activities of several CO catalysts including Pt/AC (activated carbon), Pt/TiO2, Pt/SnO2, MnO2–CuO/Al2O3, CuCl2–PdCl2–Ni/Al2O3, Au/Fe2O3, nano Au particle and Hopcalite.17,18 Noble metals such as Pt, Pd, Rh and Au show considerable activity for CO oxidation at low temperatures.19–23 However, the increasing price and decreasing storage of noble metals have limited their practical applications. Recently, transition metal oxides such as CuO, MnOx, TiO2, ZrO2 and Co3O4 have been reported as promising candidates for CO oxidation.19,21,24–30 Among these catalysts, Hopcalite has been widely utilized for low-temperature CO oxidation, because the catalyst is highly-active and cost-effective.31
However, the majority of CO catalysts have been reported to suffer from decreased oxidation activity when exposed in CO2, regardless of precious metals or transition metal compounds. Hoflund et al. reported that high-concentration CO2 would adversely affect CO oxidation activities of Au/MnOx and Pt/SnOx. They ascribed the decreased catalytic activities to CO2 retention on the surface of catalysts and covering of active sites with intermediates. They proposed to mitigate the inhabiting effect of CO2 with Fe promotion.32 Liang et al. reported that CO oxidation activity of Pd/CeO2–TiO2 would be affected by CO2 concentration and reaction temperature. They found that CO2 molecules could be dissociated into CO and activated monoatomic oxygen species at lower temperatures. This can improve the stability of the catalyst. The detrimental effect of CO2 could be deduced as the formation of carbonate and change of mechanism.33 In Benedetto's investigation, inhibiting effect had also been observed for CO oxidation over CuO/CeO2 when the catalyst was exposed in 15% CO2. They attributed the decreased CO oxidation activity in the presence of CO2 to CO2 chemisorption over the support and catalyst to form stable surface species.34 Gamarra et al. investigated the deactivating mechanism of CuO/CeO2 and Ce0.8Cu0.2O2 in the presence of CO2 using operando DRIFTS and XANES. They ascribed the catalyst deactivation to the formation of interfacial carbonates.35,36
Recently, we have evaluated CO oxidation activity of Hopcalite in the presence of CO2. Significant decay in CO oxidation activity and stability could be observed when the sample is exposed in CO2. To improve CO oxidation activity and stability, the sample is modified with KOH, and the CO oxidation performances of the modified catalyst in the presence of CO2 have also been evaluated.37 Results show that KOH modification can mitigate the inhibiting effect of CO2 on CO oxidation activity. However, the mechanisms for catalyst deactivation in the presence CO2 remain unknown. Besides, the role of KOH in the bi-functional catalyst and the mechanisms for its' positive effect on CO oxidation are not clear. In this work, experimental investigations on CO oxidation performances of Hopcalite precursor and the modified catalyst in 0.4% CO and 0.4% CO + 1.0% CO2 are conducted. Furthermore, mechanisms for catalyst deactivation in the presence of CO2 are demonstrated. The positive effect of KOH modification on CO oxidation performances are revealed. Further insights have been focused on the kinetic performances of the precursor and catalyst in 0.4% CO and 0.4% CO + 1.0% CO2. The results will lay the groundwork for large-scale application of the catalyst for post-fire cleanup in confined spaces.
2. Experimental
2.1 Catalyst preparation
Bi-functional catalyst of KOH–Hopcalite (KHC) was synthesized with the impregnation of KOH over commercial Hopcalite (HC) precursor. The precursor with an average particle size of 300 μm was purchased from Shanxi Xinhua Senquan Industrial and Trading Co., Ltd. The precursor was synthesized by an extrusion method at high pressure. KOH (99.99%) was purchased from Shanghai Jiuyi Chemical Reagent Co., Ltd. In detail, a certain gram of KOH was dissolved in 250 mL of de-ionized water to form uniform KOH solutions. Afterwards, the precursor was impregnated in KOH solutions and mixed with a magnetic stirrer overnight. The mixtures were subsequently dried at 85 °C for dehydration. Thereafter, the sample was calcined at 300 °C to obtain the desired catalyst.2.2 Catalyst characterization
N2 adsorption–desorption test was carried out on an automatic surface area and porosity analyzer Tristar II 3020M (Micromeritics, USA), to demonstrate the microscopic characteristics of the catalyst. The specific surface area and pore volume were calculated using Brunauer–Emmett–Teller (BET) and Dubinin-Radushkevich methods, respectively. The pore size distribution of the catalyst was determined by Barrett–Joyner–Halenda (BJH) method. The particle morphologies of the precursor and catalyst were obtained from field emission scanning electron microscopy (FESEM) performed on a Philips SIRION 200 (Philips, Netherlands). The SEM images were taken at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times under the accelerating voltage of 5.0 kV. X-ray photoelectron spectra (XPS) analyses were performed using an ESCALAB 250 spectrometer (Thermo-VG Scientific, UK) with Al Kα radiation (powered at 15 kV and 150 W). The spectra of Cu 2p, Mn 2p, K 2p and O 1s were recorded with constant pass energy of 30.0 eV using a 500 μm diameter analysis area. Fourier transform infrared (FT-IR) spectra of the samples were collected on a Nicolet 8700 FT-IR spectrometer (Thermo Scientific Instrument, USA) with a spectral resolution of 0.1 cm−1. The structural change of the catalyst was analyzed by X-ray diffraction (XRD). The XRD patterns were picked up on a Philips X'Pert PRO (Philips, Netherlands) using nickel-filtered Cu Kα radiation at 30 kV and 150 mA with 2θ angle ranging from 10° to 70°, wave length λ = 0.15406 nm and 0.02° sampling width. The contents of Cu, Mn and K in the catalyst were measured by inductively coupled plasma-mass spectrometry (ICP-MS) as 13.20, 30.80 and 11.97 wt%, respectively.
000 times under the accelerating voltage of 5.0 kV. X-ray photoelectron spectra (XPS) analyses were performed using an ESCALAB 250 spectrometer (Thermo-VG Scientific, UK) with Al Kα radiation (powered at 15 kV and 150 W). The spectra of Cu 2p, Mn 2p, K 2p and O 1s were recorded with constant pass energy of 30.0 eV using a 500 μm diameter analysis area. Fourier transform infrared (FT-IR) spectra of the samples were collected on a Nicolet 8700 FT-IR spectrometer (Thermo Scientific Instrument, USA) with a spectral resolution of 0.1 cm−1. The structural change of the catalyst was analyzed by X-ray diffraction (XRD). The XRD patterns were picked up on a Philips X'Pert PRO (Philips, Netherlands) using nickel-filtered Cu Kα radiation at 30 kV and 150 mA with 2θ angle ranging from 10° to 70°, wave length λ = 0.15406 nm and 0.02° sampling width. The contents of Cu, Mn and K in the catalyst were measured by inductively coupled plasma-mass spectrometry (ICP-MS) as 13.20, 30.80 and 11.97 wt%, respectively.
2.3 CO oxidation test
CO oxidation activities of the samples were evaluated in a fixed-bed reactor equipped with a gas analyzer. A total of 8–40 g of the samples were packed into the reactor. The reactor was preheated at 200 °C in pure N2 stream for 30 min to eliminate the adsorbed water vapor. Then, the reactor was cooled to ambient temperature. Simulated fire smoke gas mixtures of 0.4% CO, 1.0% CO2 and balanced air with a flow rate of 500 mL min−1 then flowed through the reactor. The temperature was then increased to a certain value with a ramping rate of 20 °C min−1. By measuring the outlet CO concentration, CO oxidation activities of the samples could be evaluated. Detailed description for the experimental setup for testing CO oxidation activities of the samples could be found in the ESI.†2.4 Activity measurement
The catalytic activities of the samples are characterized by CO conversion versus temperature. CO conversion was calculated:
 | (1) |
Besides, the temperatures required for half conversion (T50) and total CO oxidation (T100) are used to evaluate the CO oxidation performances. The time for keeping total CO conversion (t100) is applied to evaluate the stability of the catalyst.
The CO oxidation rates of the samples are calculated:38,39
| rCO = XCO/W/FCO | (2) |
3. Results and discussion
3.1 Catalytic performances
To evaluate CO oxidation activities of the precursor and catalyst, the samples are tested in 0.4% CO and 0.4% CO + 1.0% CO2, and the results are shown in Fig. 1.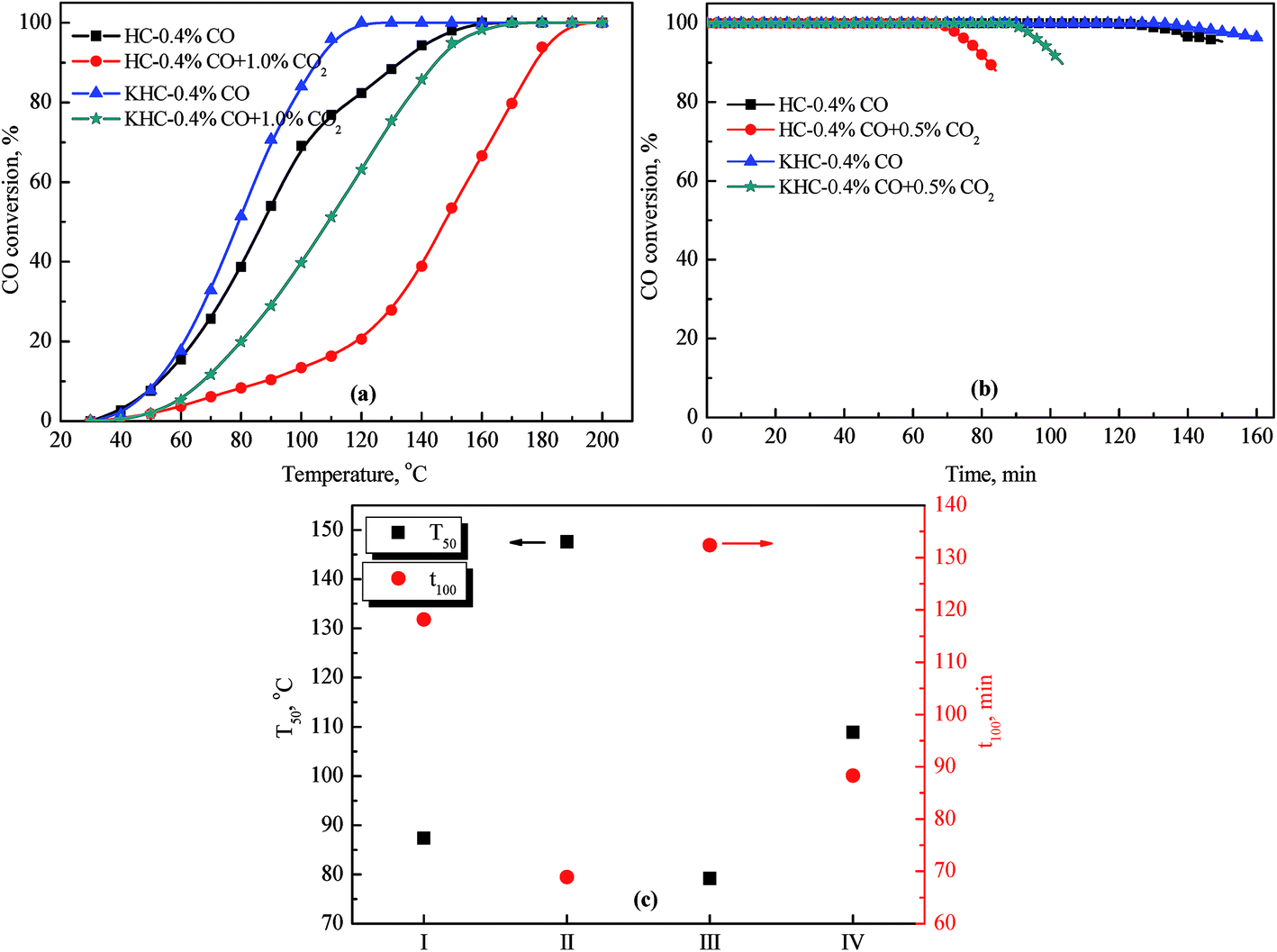 | ||
| Fig. 1 CO catalytic oxidation performances of the HC precursor and KHC catalyst in 0.4% CO and 0.4% CO + 1.0% CO2 (a) CO oxidation activity; (b) stability; (c) T50 and t100. | ||
Fig. 1(a) presents the CO conversions of the samples versus temperature in different atmospheres. CO oxidation activities of the samples increase with temperature increasing. T100 for the HC precursor in 0.4% CO and 0.4% CO + 1.0% CO2 are 160 and 190 °C, respectively. CO conversions for the precursor in 0.4% CO at the corresponding temperatures are higher than those in the presence of 1.0% CO2. This indicates that the precursor has suffered a decreased CO oxidation activity when exposed in CO2. Liang et al. reported that carbonate accumulation would occur on the surface of Pd/CeO2–TiO2, which exerted prohibitive effect on the CO oxidation activity.33 Wang et al. also indicated that CO2 would react with the active site or support on Pd–Cu catalyst to form carbonates, which showed negative effect on CO conversion.33,40,41 Hoflund et al. proposed that CO2 retention would lead to increased coverage of the surface by the intermediate species and decay of activity of CO catalyst.32 The decreased CO oxidation activity of the precursor in the presence of CO2 might be deduced as the covering of active sites and the formation of bicarbonate and carboxylate. T100 for the KHC catalyst in 0.4% CO and 0.4% CO + 1.0% CO2 are 120 and 170 °C, respectively. It is noteworthy that CO oxidation activity of KHC catalyst in 0.4% CO is higher than that of HC. This is because that the formed CO2 over KHC catalyst has been fixed as bicarbonate species, thus reducing the CO2 concentration over the active sites. CO conversion of KHC in 0.4% CO + 1.0% CO2 is higher than that of HC, which indicates that KOH modification has mitigated the inhibiting effect of CO2 on CO oxidation activity.
Fig. 1(b) presents CO conversions of the precursor and catalyst versus time in different atmospheres. CO conversion of HC in 0.4% CO decreases to 95.3% within 150 min, while that in 0.4% CO + 1.0% CO2 decreases to 87.9% within 84.1 minutes. CO conversion of KHC catalyst in 0.4% CO decreases to 96.2% in 161.3 minutes. CO conversion decreases to 89.7% within 103.5 minutes, when the catalyst is tested in 0.4% CO + 1.0% CO2. Fig. 1(c) shows that T50 for HC and KHC in 0.4% CO and 0.4% CO + 1.0% CO2 are 87.4, 147.6, 79 and 108 °C, respectively. t100 for the samples in 0.4% CO and 0.4% CO + 1.0% CO2 are determined as 118.1, 68.9, 132.4 and 88.3 min, respectively. These indicate that the presence of CO2 would adversely affect the CO oxidation activity and long-term stability of the precursor, while KOH modification could mitigate the adverse effect and improve the CO oxidation activity and stability.
3.2 Microscopic structures
Microscopic structure of the KHC catalyst is examined with N2 adsorption–desorption test. Fig. S1† presents the isotherm and pore size distribution of the catalyst. The isotherm of the catalyst belongs to type IV, as an indicative of mesoporous material.42 The slight N2 uptake under lower partial pressure is mainly depended on monolayer adsorption. The quantity of N2 adsorbed significantly increases in high partial pressure area, due to the capillary condensation of the adsorbate. Besides, obvious H3 hysteresis loop could be observed when relative pressure is higher than 0.4. This implies that the pores in the catalyst are mainly cylindrical vents lying in narrow range of radius. Pore size distribution curve of the sample shows three peaks assigned in the pore size range of 0–2, 2–5, and 10–20 nm, indicating that the pores in the sample are mainly micropores and mesopores.Table 1 shows the microstructure change of the HC precursor and KHC catalyst before and after CO oxidation in the presence of CO2. The BET surface areas of the fresh and spent precursor are 74.78 and 65.15 m2 g−1, respectively. For the fresh and spent catalyst, the BET surface areas are 67.08 and 65.42 m2 g−1, respectively. Compared to the fresh samples, the surface areas of the spent precursor and catalyst both decrease. This might be deduced as the consumption of active sites in CO oxidation process and the coverage of active sites by CO2. For the samples before and after CO oxidation, the pore volume and average pore diameter show no obvious change. Compared to the precursor, the surface area, pore volume and average pore diameter of the supported catalyst decrease, which should be ascribed to the modification of KOH.
| Samples | Surface areaa (m2 g−1) | Pore volumeb (cm3 g−1) | Average pore diameterc (nm) |
|---|---|---|---|
| a Multi-point BET surface area.b Single point adsorption total pore volume at P/P0 = 0.97.c Adsorption average pore width (4V/A by BET). | |||
| Fresh HC | 74.78 | 0.21 | 11.13 |
| Spent HC | 65.15 | 0.20 | 11.05 |
| Fresh KHC | 67.08 | 0.08 | 4.94 |
| Spent KHC | 65.42 | 0.07 | 5.24 |
3.3 Particle morphologies
The particle morphologies of the HC precursor and KHC catalyst before and after CO oxidation in the presence of CO2 are examined by SEM, and the images taken at the magnification of 10000 times are shown in Fig. 2.
 | ||
| Fig. 2 Particle morphologies of the HC precursor and KHC catalyst before and after CO oxidation in 0.4% CO + 1.0% CO2. (a) Fresh HC; (b) spent HC; (c) fresh KHC; (d) spent KHC. | ||
The morphology of the precursor shows that massive grey granules grow together to form large aggregates, which are assigned as the copper-manganese oxides compound. For the precursor after CO oxidation in the presence of CO2, the image exhibits stratified structure with obvious crystal blocks accumulated on the surface of the support. This might be deduced as the fact that the interaction between CO2 and active sites over the precursor to form bicarbonate and carboxylate has changed the surface construction. The image of the fresh catalyst shows the growth of many small white aggregates over the precursor, which should be deduced as the crystallization of KOH compounds. It can be observed that copper-manganese oxides and KOH compounds show rather uniform dispersion over the surface. The homogeneous distribution of copper-manganese oxides and uniform attachment of KOH will facilitate the catalytic process. For the catalyst after CO oxidation in the presence of CO2, many small granules are uniformly scattered on the surface. Besides, considerable mesopores could also be observed in the field of the SEM image. This is contributive to the diffusion and adsorption of the reactant gas and eventually facilitate the CO oxidation process.
3.4 FT-IR and XRD analysis
To reveal the reaction principles for catalyst deactivation and the role of KOH modification, FT-IR spectra of the samples before and after CO oxidation in different atmospheres are examined, and the results are presented in Fig. 3. The structural changes of the catalyst in different atmospheres are also examined, and the XRD patterns are presented in Fig. 4. | ||
| Fig. 3 FTIR spectra of the HC precursor and KHC catalyst. (I) Fresh HC; (II) HC in 0.4% CO; (III) HC in 0.4% CO + 1.0% CO2; (IV) fresh KHC; (V) KHC in 0.4% CO; (VI) KHC in 0.4% CO + 1.0% CO2. | ||
 | ||
| Fig. 4 XRD patterns of the KHC catalyst in different atmospheres. (I) Fresh sample; (II) in 0.4% CO; (III) in 0.4% CO + 1% CO2. ▲KOH, ■CuMn2O4, ●CuO, ♦Mn2O3, ▽KHCO3. | ||
Fig. 3 shows the FT-IR spectra of the samples before and after CO oxidation in different atmospheres. The observed characteristic peaks at 490 and 715 cm−1 are corresponding to bulk CuO phase and spinel structure of CuMn2O4, respectively. The IR bands at 530 and 1000 cm−1 are associated with the vibrational features of Mn2O3. Besides, the characteristic peaks appear at 1631 and 3480 cm−1 are attributed to carboxylate and hydroxy species, respectively. For both the precursor and catalyst in 0.4% CO and 0.4% CO + 1.0% CO2, the band at 1395 cm−1 is originated from the stretching vibration of carbonate species. This indicates that CO could be oxidized into CO2, and the adsorbed CO2 could further form carbonate species over the surface. For the modified catalyst, another peak could be observed at 862 cm−1, which should be deduced as the stretching vibration of bicarbonate species. This indicates that the formed CO2 could be captured by KOH to form bicarbonate. Besides, the carboxylate species could also be observed at 1537 cm−1, implying that the adsorbed CO2 could also be converted into carboxylate in the presence of KOH.
Fig. 4 shows the XRD patterns of the fresh and spent catalyst in 0.4% CO and 0.4% CO + 1.0% CO2. The fresh sample displays characteristic diffraction peaks of CuO and Mn2O3, indicating that CuO and Mn2O3 are the main states for Cu and Mn species, respectively. Besides, amorphous crystalline phase of CuMn2O4 are also observed. This should be deduced as the sintering of CuO and Mn2O3 in the high-temperature calcination process. The characteristic diffraction peaks with high intensity observed are assigned as KOH compounds. It can be concluded that the main phases in the sample are CuO, Mn2O3, CuMn2O4 and KOH. For the spent catalyst in 0.4% CO and 0.4% CO + 1.0% CO2, the diffraction peaks for the original KOH phase disappear. Instead, a new phase presents as KHCO3. This indicates that CO has been oxidized into CO2, and the formed CO2 has been further captured by KOH.
3.5 Reaction pathways
The mechanisms for CO oxidation process over the KHC catalyst in the presence of CO2 are proposed, on the basis of the evidence from the FT-IR and XRD results, and the schematic diagram for reaction pathways is presented in Fig. 5.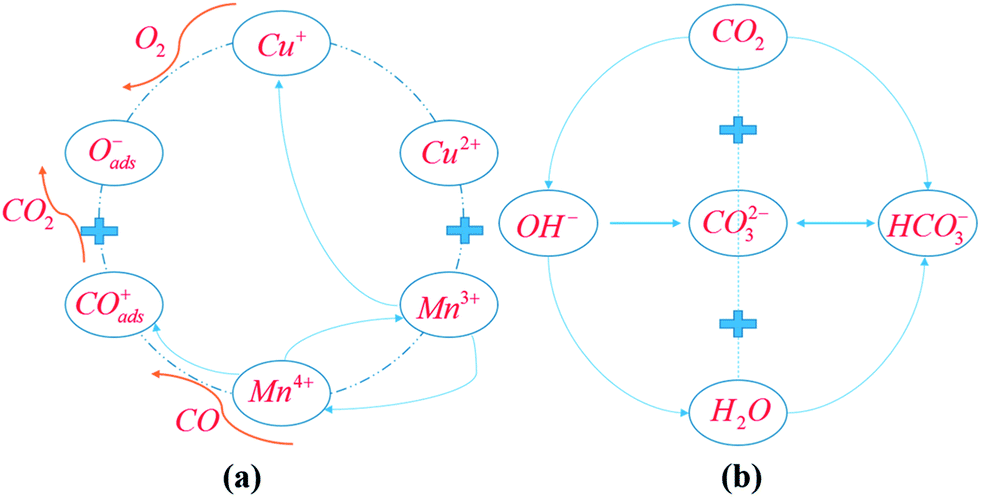 | ||
| Fig. 5 Reaction mechanisms for CO oxidation over the KHC catalyst in 0.4% CO + 1% CO2. (a) Spill-over effect for CO oxidation process; (b) proposed mechanism for CO2 sorption process. | ||
The pathways for the CO oxidation process over HC precursor could be explained by the spill-over effect proposed by Schwab and Kanungo et al.43–45 It had been verified that four distinct ions of Cu2+, Mn3+, Cu+ and Mn4+ would always be present in the spinel CuMn2O4 lattice.46 The CO catalytic oxidation process over HC mainly depends on the electronic migration between Cu and Mn cations within the spinel CuMn2O4 lattice:
| Cu2+ + Mn3+ ↔ Cu+ + Mn4+ | (3) |
In this way, polycrystalline copper manganese oxides are formed. The atoms on the surface of the oxides then provide considerable adsorption sites for the dissociated O2 and CO molecules. CO-induced reconstruction occurs on the surface of manganese oxide. CO is dissociated by Mn4+ to form CO adsorption sites:
| CO + Mn4+ → COads+ + Mn3+ | (4) |
Oxygen-induced reconstruction occurs on the surface of copper oxide. O2 is dissociated by Cu+ to form oxygen adsorption sites:
| ½O2 + Cu+ → Cu2+ + Oads− | (5) |
Then, the formed CO adsorption sites and oxygen adsorption sites migrate over the surface to combine with each other to form CO2 molecules:
| COads+ + Oads− → CO2 | (6) |
The reconstructed surfaces are then recovered through the resonance reaction system to bring the catalyst back to the initial active state, as shown in eqn (4).
The formed CO2 could be captured by KOH, to mitigate the inhibiting effect of CO2 on CO oxidation activity. On the one hand, CO2 could react with KOH to form bicarbonate species, thus reducing CO2 concentration driving force over the active sites.
| CO2 + 2OH− → CO32− + H2O | (7) |
| CO2 + CO32− + H2O → 2HCO3− | (8) |
However, the positive effect of KOH on CO2 deactivation is significant under low CO2 partial pressures. The role of KOH modification under high CO2 concentrations might not be preferable, since high-concentration CO2 means more bicarbonate species will be formed, thus reducing the active sites available for CO adsorption.
On the other hand, CO2 would react with hydroxyl to form carboxylate and oxygen adsorption sites, to replenish the depleted oxygen in the redox process. This should also be responsible for the promotion of CO oxidation activity with KOH modification.
| CO2 + OH− → COOH + Oads− | (9) |
3.6 Kinetic performances
Kinetic study on CO catalytic oxidation performances of the precursor and catalyst under different atmospheres are performed, with the weight of the samples changing from 8 to 40 g. The variation of fractional CO conversion with W/FCO ratio under typical temperatures are plotted in Fig. 6. | ||
| Fig. 6 Variation of fractional CO conversion with the W/FCO ratio (a) HC in 0.4% CO; (b) HC in 0.4% CO + 1.0% CO2; (c) KHC in 0.4% CO; (d) KHC in 0.4% CO + 1.0% CO2. | ||
For both the precursor and catalyst, the fractional CO conversions in different atmospheres increase linearly with W/FCO ratio, which implies that more CO could be converted into CO2. As the weight of the sample increases, the active sites available for CO adsorption increases, and CO oxidation activity is enhanced. When the W/FCO ratio is fixed, CO conversion increases with temperature increasing, as an indicative of enhanced CO oxidation activity at higher temperatures. With temperature increasing, more active sites are available for CO adsorption. This will lead to an increased turnover efficiency (TOF) and higher CO conversion.47 The CO oxidation rates of the samples under different temperatures in 0.4% CO and 0.4% CO + 1.0% CO2 are calculated from the slopes of the linear plots, and the results are plotted in Fig. 7.
 | ||
| Fig. 7 Arrhenius plot of CO oxidation for the HC precursor and KHC catalyst in 0.4% CO and 0.4% CO + 1.0% CO2. | ||
Fig. 7 presents the change of CO oxidation rates with temperature for the samples under different atmospheres. The variation of CO oxidation rates with temperature is found to follow the Arrhenius-type expression, and the plot satisfies a linear relationship. The activation energies for the CO oxidation processes of the samples in different atmospheres are determined from the slopes of the fitting lines. The activation energies for CO oxidation processes over HC in 0.4% CO and 0.4% CO + 1.0% CO2 are 34.59 and 60.69 kJ mol−1, respectively. Those for the KHC catalyst in the two atmospheres are calculated as 22.95 and 27.77 kJ mol−1, respectively. The activation energy for CO oxidation process over HC in 0.4% CO + 1.0% CO2 is much higher than that in 0.4% CO, indicating that the precursor would suffer from deactivation in the presence CO2. The CO oxidation process over KHC in 0.4% CO + 1.0% CO2 shows a rather lower activation energy than that for HC, which implies that KOH modification has mitigated the inhibiting effect of CO2. The activation energy for CO oxidation process over KHC in 0.4% CO is rather lower than that for HC. This indicates that KOH modification has promoted the CO catalytic oxidation activity of the HC precursor.
It has been identified that the catalytic activity, heat of adsorption and surface coverage are closely related with each other.48 Due to the surface heterogeneity, heat of adsorption would decrease with the increase of surface coverage. For CO oxidation processes of HC and KHC in the presence of CO2, the competitive adsorption between CO and CO2 has resulted in less active sites available for CO adsorption. This means that the surface coverage for CO adsorption will decrease, while the heat of adsorption will increase. The temperature required for higher CO oxidation activity will increase. This is the reason why the temperatures and activation energies required for total CO conversion of HC precursor and KHC catalyst in the presence of 0.4% CO + 1.0% CO2 are higher than those in 0.4% CO.
4. Conclusions
A bi-functional catalyst of KOH–Hopcalite is prepared by impregnation of KOH over commercially available Hopcalite precursor. CO oxidation performances of the precursor and catalyst are evaluated in the presence of CO2. CO oxidation activity of the precursor would decrease when exposed in CO2, due to the covering of active sites and the formation of bicarbonates and carboxylate. KOH modification is found to mitigate the adverse effect of CO2 and improve the CO oxidation activity and stability. The CO oxidation process over the catalyst is depended on the electronic migration between Cu and Mn cations within the spinel CuMn2O4 lattice. KOH modification plays an important role in the multi-phase catalytic system, namely, to capture the formed CO2 to reduce the CO2 concentration over the active sites, and to provide hydroxyl to be dissociated as oxygen adsorption sites to replenish the depleted oxygen in the redox process. These will eventually facilitate the CO oxidation process for the bi-functional catalyst in the presence of CO2. The apparent activation energy for the CO oxidation process of the bi-functional catalyst in the presence of CO2 is determined as 27.77 kJ mol−1, which is much lower than that of 60.69 kJ mol−1 for the precursor. The modified catalyst could be potentially utilized as an effective option for post-fire cleanup and atmospheric recovery in confined spaces.Acknowledgements
Financial support from the Joint Fund of the National Natural Science Foundation of China and Shanxi Province (U1510129), the National Natural Science Foundation of China (51323010 and 51206155) and the Fundamental Research Funds for the Central Universities (WK2320000034) is sincerely acknowledged. The authors also wish to acknowledge Dr Yanming Ding for the English editing for this article.Notes and references
- Y. Hasegawa, K. Fukumoto, T. Ishima, H. Yamamoto, M. Sano and T. Miyake, Appl. Catal., B, 2009, 89, 420–424 CrossRef CAS.
- G. B. Hoflund, S. D. Gardner, D. R. Schryer, B. T. Upchurch and E. J. Kielin, Appl. Catal., B, 1995, 6, 117–126 CrossRef CAS.
- A. A. Mirzaei, H. R. Shaterian, R. W. Joyner, M. Stockenhuber, S. H. Taylor and G. J. Hutchings, Catal. Commun., 2003, 4, 17–20 CrossRef CAS.
- T. Cheng, Z. Fang, Q. Hu, K. Han, X. Yang and Y. Zhang, Catal. Commun., 2007, 8, 1167–1171 CrossRef CAS.
- A. Luengnaruemitchai, D. T. K. Thoa, S. Osuwan and E. Gulari, Int. J. Hydrogen Energy, 2005, 30, 981–987 CrossRef CAS.
- E. C. Njagi, C. H. Chen, H. Genuino, H. Huang and S. L. Suib, Appl. Catal., B, 2010, 99, 103–110 CrossRef CAS.
- G. J. Hutchings, A. A. Mirzaei, R. W. Joyner, M. R. H. Siddiqui and S. H. Taylor, Catal. Lett., 1996, 42, 21–24 CrossRef CAS.
- S. H. Taylor and C. Rhodes, Catal. Lett., 2005, 101, 31–33 CrossRef CAS.
- Y. Jia, Y. Liu, W. Liu and Z. Li, Sep. Purif. Technol., 2014, 130, 65–73 CrossRef CAS.
- L. Mallon, A review of the currently available methods for ambient temperature carbon monoxide removal in a disabled royal navy submarine, Proceedings of the 38th International Conference on Environmental Systems, San Francisco, California, 2008, DOI:10.4271/2008-01-2126.
- F. Sribnik, P. J. Birbara, J. J. Faszcza and T. A. Nalette, SAE Trans., 1990, 99, 1145–1153 Search PubMed.
- M. Karanjika, Low temperature oxidation of carbon monoxide using microfibrous entrapped catalysts for fire escape mask application, Auburn University, 2005 Search PubMed.
- A. Gold, W. A. Burgess and E. V. Clougherty, Am. Ind. Hyg. Assoc. J., 1978, 39, 534–539 CrossRef CAS PubMed.
- J. B. Terrill, R. R. Montgomery and C. F. Reinhardt, Science, 1978, 200, 1343–1347 CAS.
- S. Royer and D. Duprez, ChemCatChem, 2011, 3, 24–65 CrossRef CAS.
- E. Jin, L. He, Y. Zhang, A. R. Richard and M. H. Fan, RSC Adv., 2014, 4, 48901–48904 RSC.
- T. Nalette, C. Eldridge, P. Yu, G. Alptekin and J. Graf, Advanced catalysts for the ambient temperature oxidation of carbon monoxide and formaldehyde, Proceedings of the AIAA 40th International Conference on Environmental Systems, Barcelona, Spain, 2010, DOI:10.2514/mices10.
- B. Luna, G. Somi, J. P. Winchester, J. Grose, L. Mulloth and J. L. Perry, Evaluation of commercial off-the-shelf sorbents and catalysts for control of ammonia and carbon monoxide, Proceedings of the AIAA 40th International Conference on Environmental Systems, Barcelona, Spain, 2010, DOI:10.2514/mices10.
- N. Li, Q. Y. Chen, L. F. Luo, W. X. Huang, M. F. Luo, G. S. Hu and J. Q. Lu, Appl. Catal., B, 2013, 142, 523–532 CrossRef.
- Y. Li, Y. Yu, J. G. Wang, J. Song, Q. Li, M. Dong and C. J. Liu, Appl. Catal., B, 2012, 125, 189–196 CrossRef CAS.
- A. Satsuma, K. Osaki, M. Yanagihara, J. Ohyama and K. Shimizu, Appl. Catal., B, 2013, 132, 511–518 CrossRef.
- L. C. Wang, Q. Liu, X. S. Huang, Y. M. Liu, Y. Cao and K. N. Fan, Appl. Catal., B, 2009, 88, 204–212 CrossRef CAS.
- J. Huang, L. C. Wang, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Appl. Catal., B, 2011, 101, 560–569 CrossRef CAS.
- Y. Hasegawa, K. Fukumoto, T. Ishima, H. Yamamoto, M. Sano and T. Miyake, Appl. Catal., B, 2009, 89, 420–424 CrossRef CAS.
- Z. Boukha, J. L. Ayastuy, A. Iglesias-González, B. Pereda-Ayo, M. Gutiérrez-Ortiz and J. R. González-Velasco, Appl. Catal., B, 2014, 160, 629–640 CrossRef.
- T. J. Clarke, T. E. Davies, S. A. Kondrat and S. H. Taylor, Appl. Catal., B, 2015, 165, 222–231 CrossRef CAS.
- Y. Feng, L. Li, S. Niu, Y. Qu, Q. Zhang, Y. Li and J. Shi, Appl. Catal., B, 2012, 111, 461–466 CrossRef.
- Y. Lou, L. Wang, Z. Zhao, Y. Zhang, Z. Zhang, G. Lu and Y. Guo, Appl. Catal., B, 2014, 146, 43–49 CrossRef CAS.
- J. Li, G. Z. Lu, G. S. Wu, D. S. Mao, Y. L. Guo, Y. Q. Wang and Y. Guo, RSC Adv., 2013, 3, 12409–12416 RSC.
- A. Alvarez, S. Ivanova, M. A. Centeno and J. A. Odriozola, Appl. Catal., A, 2012, 431, 9–17 CrossRef.
- Y. Li, H. Peng, X. Xu, Y. Peng and X. Wang, RSC Adv., 2015, 5, 25755–25764 RSC.
- G. B. Hoflund, S. D. Gardner, D. R. Schryer, B. T. Upchurch and E. J. Kielin, Langmuir, 1995, 11, 3431–3434 CrossRef CAS.
- F. Liang, H. Zhu, Z. Qin, G. Wang and J. Wang, Catal. Commun., 2009, 10, 737–740 CrossRef CAS.
- A. Di Benedetto, G. Landi, L. Lisi and G. Russo, Appl. Catal., B, 2013, 142, 169–177 CrossRef.
- D. Gamarra and A. Martínez-Arias, J. Catal., 2009, 263, 189–195 CrossRef CAS.
- D. Gamarra, M. Fernández-García, C. Belver and A. Martínez-Arias, J. Phys. Chem. C, 2010, 114, 18576–18582 CAS.
- Y. Guo, C. Zhao, C. Li and S. Lu, Low Temperature CO Catalytic Oxidation over KOH-Hopcalite Mixtures and In situ CO2 Capture from Fire Smoke, The 10th Asia-Oceania Symposium for Fire Science and Technology, Tsukuba, Japan, 2015 Search PubMed.
- S. A. Singh and G. Madras, Appl. Catal., A, 2015, 504, 463–475 CrossRef CAS.
- V. M. Shinde and G. Madras, Catal. Sci. Technol., 2012, 2, 437–446 CAS.
- F. Liang, K. Zhao, H. Zhang, Y. Dong, T. Wang and D. He, Chem. Eng. J., 2014, 242, 10–18 CrossRef.
- Y. Shen, G. Lu, Y. Guo, Y. Wang, Y. Guo and X. Gong, Catal. Today, 2011, 175, 558–567 CrossRef CAS.
- S. J. Gregg and K. S. W. Sing, Adsorption, surface area, and porosity, Academic Press, London, 2nd edn, 1995 Search PubMed.
- F. C. Buciuman, F. Patcas and T. Hahn, Chem. Eng. Process., 1999, 38, 563–569 CrossRef CAS.
- G. M. Schwab and S. B. Kanungo, Z. Phys. Chem., 1977, 107, 109–120 CrossRef CAS.
- S. B. Kanungo, J. Catal., 1979, 58, 419–435 CrossRef CAS.
- A. Waskowska, L. Gerward, J. S. Olsen, S. Steenstrup and E. Talik, J. Phys.: Condens. Matter, 2001, 13, 2549–2562 CrossRef CAS.
- L. Wang, W. Wang, Y. Zhang, Y. Guo, G. Lu and Y. Guo, Catal. Today, 2015, 242, 315–321 CrossRef CAS.
- T. B. Grimley and S. M. Walker, Surf. Sci., 1969, 14, 395–406 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra23806d |
| This journal is © The Royal Society of Chemistry 2016 |