
Complexing agent study for environmentally friendly silver electrodeposition(II): electrochemical behavior of silver complex
Anmin Liu,
Xuefeng Ren,
Jie Zhang,
Deyu Li and
Maozhong An*
State Key Laboratory of Urban Water Resource and Environment, School of Chemical Engineering and Technology, Harbin Institute of Technology, Harbin, 150001, China. E-mail: mzan@hit.edu.cn; Fax: +86-451-86418616; Tel: +86-451-86418616
First published on 7th January 2016
Abstract
Electrochemical behaviors of a silver complex in an environmentally friendly silver plating bath with 5,5-dimethylhydantoin (DMH) and nicotinic acid (NA) as complexing agents were investigated in this paper. Like the cyanide based silver electroplating bath, silver deposits with smooth and compact morphologies, as well as high purity could be obtained from the studied DMH and NA based silver electroplating bath. The electrochemical behaviors of the silver complex in this cyanide-free silver electroplating were studied by cyclic voltammetry with different sweep rates to investigate the discharge process of the silver plating bath, the transfer coefficients and diffusion coefficient of the silver complex in this bath. The results of the chronoamperometry on a glass carbon electrode indicate that the nucleation processes of silver from the bath is a three-dimensional progressive nucleation process, and the growth of silver nucleation and the nucleation rate of silver deposit in the DMH and NA based silver plating bath were highly dependent on the applied potential.
1. Introduction
Silver deposits are widely applied in many industrial areas owing to its excellent physical and chemical properties.1–4 Electrodeposition is one of the most important and low cost methods to get excellent silver deposits for a variety of applications, in which the cyanide based electroplating baths have been used for more than 100 years to obtain compact, smooth, and adhesive silver deposits with the most consistent quality at the lowest cost.5–9 Unfavorably, as one of the most poisonous chemicals, cyanide brings extremely high hazardous risks to human health and the environment.10 Furthermore, with the increasing pressure of environmental protection, the disposal of exhausted electroplating baths and wastewater are becoming increasingly difficult and expensive.11,12In order to overcome these challenges and eliminate cyanide-based baths, a number of attempts have been made in past few years to develop cyanide-free silver electroplating baths. Among them, complexing agents for cyanide-free silver electroplating have been widely investigated owing to their important role during the electroplating process. Complexing agents for cyanide-free silver electroplating baths, such as thiosulfate,13–16 uracil,17 ammonia,18,19 sodium citrate,20 HEDTA,21 2-hydroxypyridine,22,23 and ionic liquids,10,24 have been proposed. Nevertheless, except for a few successful cases, most of these baths still suffer from problems of instability of electroplating baths and low deposit quality, including severe adhesion and inferior morphologies. Consequently, more effort should be directed toward this area to find a comparable and environmentally friendly alternative to cyanide-based silver electroplating baths.
5,5-Dimethylhydantoin (DMH) and nicotinic acid (NA), two heterocyclic structured organics, were selected as complexing agents for cyanide-free silver electroplating in our study.25,26 Compared to other reported complexing agents used for cyanide-free silver electroplating baths, the DMH and NA molecules have good solubility and stability in alkaline solution in a large temperature range, and act as more stable complexing agents for silver(I).27 In order to improve the practicability of the introduced bath and silver deposit in more and more areas, a variety of DMH and NA based cyanide-free silver electroplating baths were developed.25,26,28
Besides the application of silver deposits in electronics or other industries, the investigation of the electrochemical behavior of the silver complex in an environmentally friendly silver plating bath is of great importance. The cathodic deposition process29–31 and the nucleation and growth mechanisms22,32–36 were the most important properties to study the electrochemical behaviors of the silver complex in the DMH and NA based cyanide-free silver plating baths.
In the present work, the performances of the silver deposit obtained from the DMH and NA based silver electroplating bath were determined by experiments and compared with that from the cyanide based bath. As with the cyanide based silver electroplating bath, a mirror bright silver deposit, with excellent leveling capability, smooth and compact morphologies, and high purity could be obtained from the studied DMH and NA based silver electroplating bath. The cathodic deposition process of the silver complex in the investigated cyanide-free silver electroplating bath was investigated by cyclic voltammetry. Chronoamperometry was employed to explore the initial process of the silver electrodeposition, including the nucleation and growth mechanisms. Cathodic polarization was demonstrated by potentiodynamic cathodic polarization curves on the Pt rotating disk electrode to study the activation energy of the silver electrodeposition process.
2. Experimental
All silver electroplating baths used in this work were prepared using analytical grade reagents and deionized water. The introduced silver electroplating bath was prepared by adding 0.075 M AgNO3 solution into an electrolyte containing 0.7 M DMH, 0.7 M NA, and 1.3 M K2CO3, the pH value of the electroplating bath was adjusted to 10.0–14.0 with KOH solution after the addition of AgNO3 solution. Silver electroplating experiments were conducted under galvanostatic conditions (at 0.8 A dm−2) in a cell employing a copper sheet as the cathodic substrate.Field emission scanning electron microscopy (FE-SEM) was employed to characterize the surface morphologies of the silver deposits obtained from the introduced silver electroplating bath and the cyanide based one. The detection of impurities in these two studied silver deposits was performed by auger electron spectroscopy (AES), and the original surface and a 1.0 nm depth of silver deposit were studied to analyze the contents of Ag, C, N, and O to investigate the impurities in the silver deposits from the introduced electroplating bath and cyanide based electroplating bath.
All electrochemical measurements were performed in a typical three-electrode cell connected with an electrochemical workstation. A platinum foil electrode and a mercuric oxide electrode (Hg/HgO) were employed as the counter electrode (CE) and the reference electrode (RE), respectively. A glassy carbon electrode (GCE) with a diameter of 3 mm was used for cyclic voltammetry (CV) and chronoamperometry (I–t) measurements.
3. Results and discussion
3.1. Morphology of silver deposits
The surface morphology is a very important consideration for most silver deposit applications. Fig. 1 displays the top view SEM images of the silver deposits obtained from the cyanide-based electroplating bath and the cyanide-free electroplating bath introduced in this work. | ||
| Fig. 1 Top view SEM images of full bright silver deposits obtained from different silver electroplating baths, (a) and (c) from the cyanide based silver electroplating bath, (b) and (d) from the DMH and NA based silver electroplating bath. | ||
From comparison of the low definition images (Fig. 1(a) and (b)), the silver deposits obtained from the two baths have the same flatness. On the other hand, no difference can be observed between the high definition images (Fig. 1(c) and (d)), as the grain size and compactness are also the same. Therefore, the silver deposits from these two electroplating baths have the same morphology. It can therefore be concluded that mirror bright silver deposits on copper substrates, with excellent leveling capability, smooth and compact morphologies as well as tiny silver particles, can be obtained from the investigated DMH and NA based cyanide-free silver electroplating bath.
3.2. Purity of silver deposits
Impurity, the interfused organic complexing agents or additives in the deposits, is another key consideration in silver electrodeposition because it seriously affects the properties and quality of the resulting silver deposits. When applied in microelectronics or decorative industries, the vitally important anti-tarnish abilities and electrical properties of the silver deposits are severely compromised by the interfused impurities.37–39 In order to investigate the impurities in the silver deposits from the DMH and NA based electroplating bath, compared with that from the cyanide based electroplating bath, AES analysis was carried out to examine the composition and proportion of the atoms in the silver deposits.Fig. 2 displays the composition of the original surface and a 1.0 nm depth of silver deposit obtained from the introduced cyanide-free electroplating bath and the cyanide one. The proportions of atoms at the original surface and a 1.0 nm depth of silver deposit measured by AES are shown in Table 1.
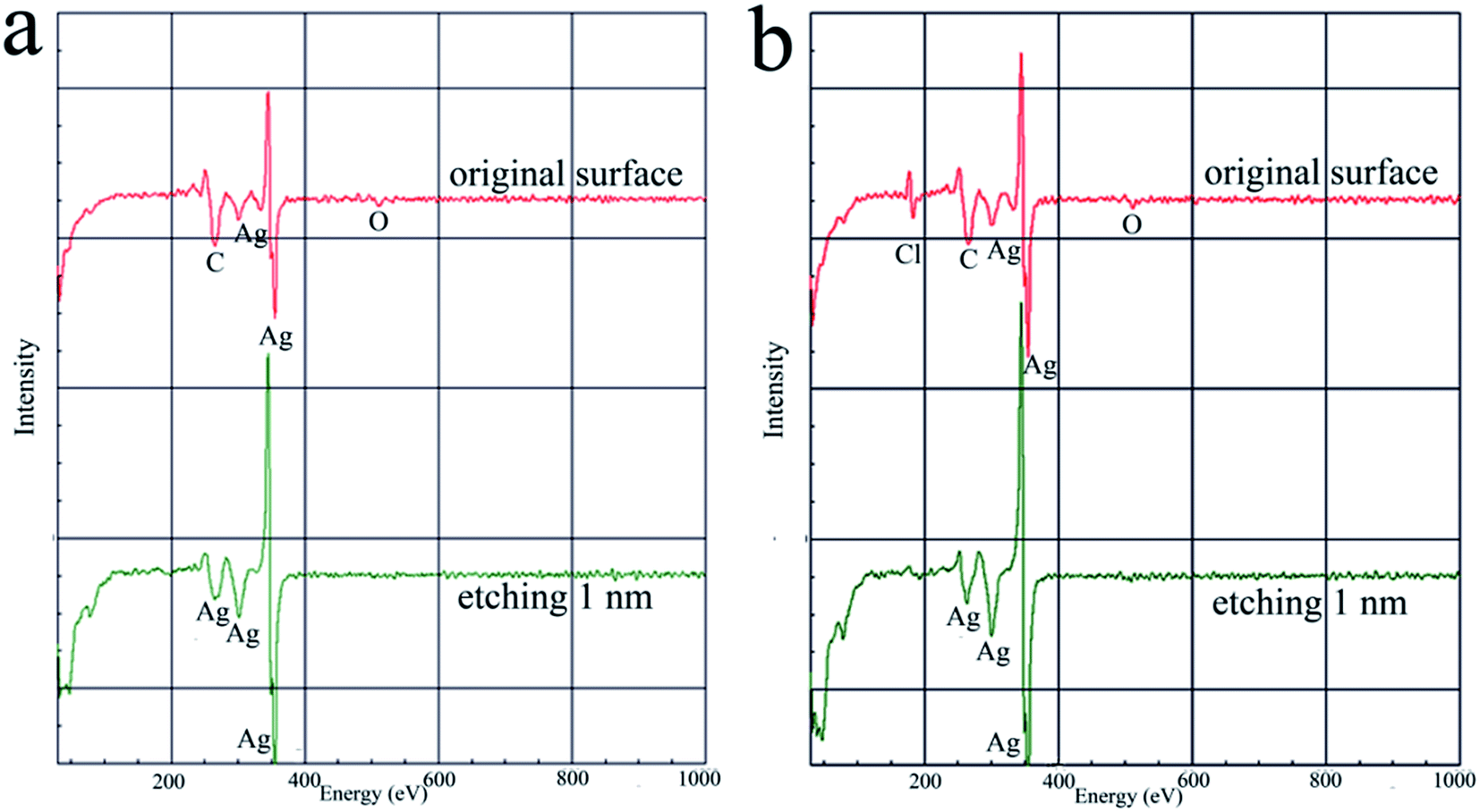 | ||
| Fig. 2 AES spectra of the original surface and a 1.0 nm depth of silver deposits, (a) from the cyanide based silver electroplating bath, (b) from the DMH and NA based silver electroplating bath. | ||
| Ag | C | O | Cl | ||
|---|---|---|---|---|---|
| Cyanide deposit | Original surface | 31.1 | 64.6 | 4.3 | 0 |
| Etching 1 nm | 100.0 | 0 | 0 | 0 | |
| Cyanide-free deposit | Original surface | 37.2 | 52.4 | 4.4 | 6.0 |
| Etching 1 nm | 100.0 | 0 | 0 | 0 | |
As displayed in Fig. 2(a), Ag, C, N, and O were present on the original surface of deposit from the cyanide based silver electroplating bath. Contrastingly, besides the signals of Ag, C, N, and O, a signal for Cl could be detected on the original surface of deposit from the introduced cyanide-free silver electroplating bath, as shown in Fig. 2(b). However, the signal for Cl could not represent the contaminant on the silver surface as no Cl was used in the bath and it was only in the top 1.0 nm.
On the other hand, no signals for C, N, O, and Cl could be detected at the 1.0 nm depth of silver deposit at the sensitivity limit of AES, indicating that all these organic impurities only adsorbed on the surface of cyanide and cyanide-free based silver deposits, and the purity of the silver deposit was very high.
With high purity, the introduced cyanide-free silver deposit has excellent anti-tarnish abilities and electrical properties. Surface adsorption of organic impurities could not influence the conductivity of the bulk silver since the surface adsorption layer was only a single molecular layer. Thus, the silver deposit obtained from the introduced bath could be used in microelectronics and decorative applications owing to its high purity, excellent anti-tarnish abilities and electrical properties.
3.3. Study of discharge process
The cathodic discharge process of the silver complex in the investigated cyanide-free silver electroplating bath was investigated by cyclic voltammetry (CV). The potential scan started at the open circuit potential (OCP) toward the negative direction with a sweep rate of 20 mV s−1 from −1.35 V to 1.10 V, as shown in Fig. 3. | ||
| Fig. 3 Cyclic voltammograms with a sweep rate of 20 mV s−1 on the GCE of the investigated silver electroplating bath. | ||
In the forward scan, as the scanning potential reaches a relatively negative value (at point H) the cathodic current begins increasing and results in a cathodic peak (at point B), then the cathodic current decreases as the scanning potential becomes more negative for the diffusion control until the current increase of the hydrogen evolution area (at point C). In the positive scanning direction, the cathodic current decreases slowly and makes the reverse curve intersect the forward cathodic current curve at point G, resulting in forming a “hysteresis loop”, a characteristic of the three-dimensional (3D) nucleation and growth of silver on the GCE surface.40–43 In addition, an anodic dissolution peak of silver deposit (at point D) and an oxygen evolution area (at point E) emerged during the positive direction scanning.
The CVs displayed in Fig. 4 at various sweep rates on 3 mm GCE were obtained to study the electrochemical behaviors of the silver complex in the cathodic deposition process, including the control step of silver electrodeposition, the transfer coefficient, and the diffusion coefficient.
 | ||
| Fig. 4 Cyclic voltammograms with different sweep rates on the GCE of the studied silver electroplating bath. | ||
As displayed in Fig. 4, the cathodic peak (B) current density jp was different to the anodic peak (A) current density, it increases with the increase of the sweep rate and the cathodic peak (B) potential (Ep) moved more to the negative side as the sweep rate increased. These results indicate that the deposition process of the silver complex in the DMH and NA based bath is irreversible.
For the irreversible cathodic electrode process, the following equations can be used to study the kinetic features of the silver deposition process.44
| |Ep − Ep/2| = 1.857RT/(αnF) | (1) |
| jp = 0.4958(nF)3/2(αDv)1/2(RT)−1/2c | (2) |
| v/mV s−1 | jp/A dm−2 | Ep/V | ∣Ep − Ep/2∣/V | α |
|---|---|---|---|---|
| 10 | 0.368 | −0.770 | 0.136 | 0.386 |
| 20 | 0.568 | −0.805 | 0.105 | 0.499 |
| 30 | 0.754 | −0.832 | 0.080 | 0.656 |
| 40 | 0.981 | −0.854 | 0.069 | 0.761 |
| 50 | 1.043 | −0.870 | 0.068 | 0.772 |
| Average | 0.615 |
It can be seen from Fig. 5(a) that peak potential Ep plotted vs. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) v gives a linear response in the silver electroplating bath, which is characteristic of an irreversible electrode process for the silver electrodeposition.
v gives a linear response in the silver electroplating bath, which is characteristic of an irreversible electrode process for the silver electrodeposition.
 | ||
| Fig. 5 (a) The peak potential (Ep) of CV with various sweep rates versus log v of the silver electroplating bath and (b) the peak current density (jp) of CV with various sweep rates versus v1/2 of the silver electroplating bath. | ||
Fig. 5(b) displayed that the jp versus the square root of the scan rate (v1/2) of the silver plating bath possesses a linear response in the investigated silver plating bath, indicating that the discharge process of silver electrodeposition is diffusion controlled in the silver electroplating bath. This proves that silver electrodeposition in the studied bath is a typical irreversible process under diffusion control. The diffusion coefficient D in the silver plating bath containing 0.075 M Ag complex was calculated to be 7.27 × 10−8 cm2 s−1 from Fig. 5(b) and eqn (2).
3.4. Nucleation mechanism
Early stages of the silver electrocrystallisation are important as they can determine the final morphology of the macroscopic and microcosmic structure of the silver deposit, such as the brightness and smoothness. The nucleation rate and the number of crystallites formed can strongly depend on the overpotential. It is therefore important to establish the exact relationship between the overpotential and the nucleation rate as well as the number of crystallites. The classical electrochemical technique, chronoamperometry, is based on current transient measurements and has been extensively used to probe nucleation and growth phenomena in electrodeposition processes. The chronoamperometry investigation of the silver plating bath in this work is displayed in Fig. 6.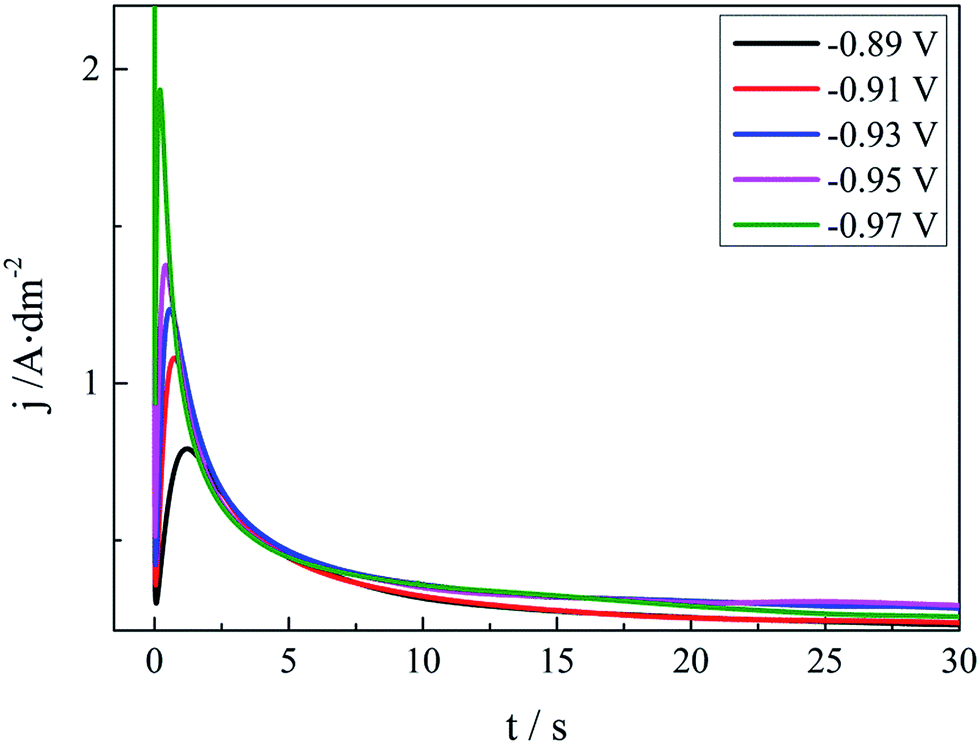 | ||
| Fig. 6 Current transients at various applied potentials on the GCE of the DMH and NA based silver electroplating bath. | ||
As shown in Fig. 6, all these I–t curves obtained from the DMH and NA based silver plating bath have a common feature. At the very beginning of the potential step, the current declines sharply owing to the double-layer charging. Then the current increases and reaches the maximum current peak at about 1.0 s to 2.0 s, and decreases subsequently after 2.0 s because of the nucleation and the growth of the silver deposit and the variation of the diffusion layer. The Scharifer and Hills (SH)43,45,46 model was employed to analyze the current transients measured in the present work. According to the SH models,33,45,47,48 the relationship of current density (j) and time (t) can be expressed by eqn (3) and (4) for 3D instantaneous nucleation and progressive nucleation, respectively. All experimental current transients were transformed into non-dimensional curves of (j/jm)2 vs. (t/tm), and the curves were compared with theoretical current transients curves, as shown in Fig. 7.
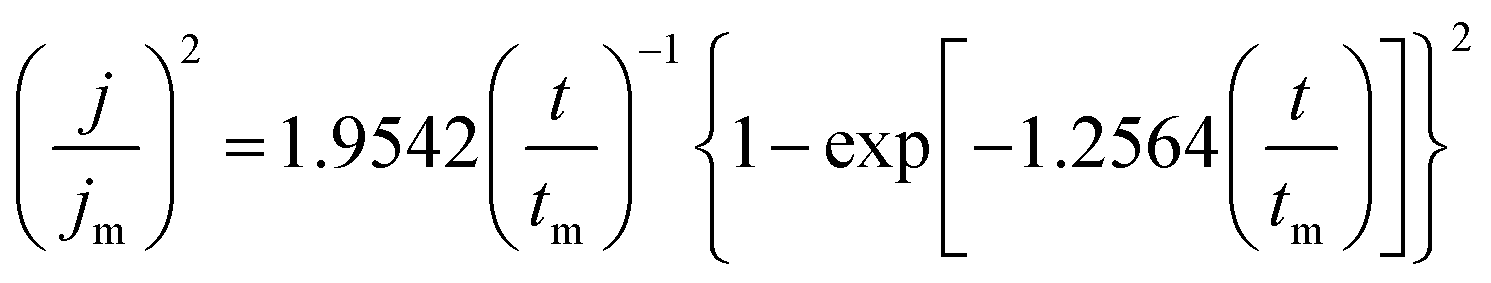 | (3) |
 | (4) |
 | ||
| Fig. 7 Non-dimensional plots of instantaneous and progressive nucleation models with three-dimensional nuclei growth and experimental curves of the silver electroplating bath. | ||
As displayed in Fig. 7, it is clear that the experimental curves of (j/jm)2 vs. (t/tm) at all potentials are very close to the theoretical curves of progressive nucleation. This indicates that the electrodeposition of silver on GCE from the electroplating bath is a progressive nucleation process.
For a 3D nucleation process, a linear relationship of j vs. tx in the rising part of the current transients can be used to reveal the electrocrystallization mechanism.29,49 It is obvious that in our case, x = 3/2 makes j vs. tx linear, as displayed in Fig. 8. Therefore, it is reasonable that the silver electrocrystallization mechanism in this work is explained by the 3D nucleation progressive model deduced from the diffusion-controlled growth model, agreeing well with the conclusions of CV measurement.
 | ||
| Fig. 8 Fits for the rising part of these transients to the progressive nucleation model of the silver electroplating bath. | ||
In the progressive nucleation process, eqn (5)–(8) are always used for the study of diffusion coefficient, the nuclear number density, and nucleation rate constant.22,29,45,49–51
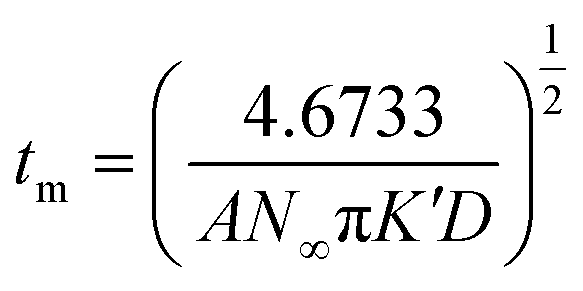 | (5) |
 | (6) |
| jm2tm = 0.2598(nFc)2D | (7) |
 | (8) |
The maximum value of the number of nuclei (saturation number Ns) and the vertical growth of silver nucleation (kv) can be determined by eqn (9)–(11).
 | (9) |
 | (10) |
 | (11) |
Parameters according to eqn (5)–(11) obtained from chronoamperometry at GCE in the studied cyanide-free silver plating bath with different applied potentials are summarized in Table 3.
| E/V | jm/A dm−2 | tm/s | D/10−8 cm2 s−1 | AN∞/108 cm−2 s | Ns/106 cm−2 | kv/10−8 mol s−1 cm−2 |
|---|---|---|---|---|---|---|
| −0.89 | 0.792 | 1.198 | 5.522 | 0.320 | 7.030 | 8.207 |
| −0.91 | 1.082 | 0.744 | 6.397 | 0.716 | 9.767 | 11.212 |
| −0.93 | 1.236 | 0.569 | 6.387 | 1.227 | 12.796 | 12.808 |
| −0.95 | 1.376 | 0.423 | 5.886 | 2.409 | 18.678 | 14.259 |
| −0.97 | 1.935 | 0.202 | 5.557 | 11.189 | 41.429 | 20.052 |
As displayed in Table 3, the diffusion coefficient calculated using the data of I–t measurements was almost the same as that from the CVs in Fig. 4 and 5.
It is observed that nucleation rate, AN∞, and saturation nuclei number, Ns, present an obvious increase as the applied potentials move negatively. Indicating that with a bigger overpotential, the nucleation rate can be expressed. The vertical growth of silver nucleation was significantly enhanced by the increase of the cathodic overpotential. As displayed in Fig. 9 and 10, lgkv − E and lnNs − E give linear responses in the investigated silver plating bath, proving that the growth of silver nucleation (kv) and the nucleation rate of DMH and NA based silver plating bath are highly dependent on the applied potential.
 | ||
| Fig. 9 lgkv as a function of E for silver electroplating bath in DMH and NA based bath. | ||
 | ||
| Fig. 10 lnNs as a function of E for silver electroplating bath in DMH and NA based bath. | ||
4. Conclusions
In the cyanide-free silver electroplating bath with DMH and NA as complexing agents, as with the cyanide based silver electroplating bath, silver deposits with smooth and compact morphologies as well as high purity could be obtained.Based on the study of CV measurements, the deposition process of the silver complex in the DMH and NA based bath is irreversible. The transfer coefficient was calculated as 0.615 in the silver plating bath containing 0.075 M Ag complex and the diffusion coefficient, D, in the silver plating bath was calculated to be 7.27 × 10−8 cm2 s−1. In the study of the nucleation process of the silver deposition, it is reasonable that the silver electrocrystallization mechanism in this work is explained by the 3D nucleation progressive model. The diffusion coefficient calculated using the data of I–t measurements was almost similar to that from the CVs. It was observed that nucleation rate, AN∞, and saturation nuclei number, Ns, present an obvious increase as the applied potentials move negatively. The growth of silver nucleation (kv) and the nucleation rate of silver deposit in DMH and NA based silver plating bath were highly dependent on the applied potential.
Acknowledgements
Financial support from the State Key Laboratory of Urban Water Resource and Environment (Harbin Institute of Technology) (2015DX09) for this work is gratefully acknowledged.References
- R. Zhang, W. Lin, K. Lawrence and C. P. Wong, Int. J. Adhes. Adhes., 2010, 30, 403–407 CrossRef CAS.
- Y. Shacham-Diamand, A. Inberg, Y. Sverdlov and N. Croitoru, J. Electrochem. Soc., 2000, 147, 3345–3349 CrossRef CAS.
- N. Fishelson, A. Inberg, N. Croitoru and Y. Shacham-Diamand, Microelectron. Eng., 2012, 92, 126–129 CrossRef CAS.
- R. Manepalli, F. Stepniak, S. A. Bidstrup-Allen and P. A. Kohl, IEEE Trans. Adv. Packag., 1999, 22, 4–8 CrossRef CAS.
- K. Márquez, G. Staikov and J. W. Schultze, Electrochim. Acta, 2003, 48, 875–882 CrossRef.
- B. C. Baker, M. Freeman, B. Melnick, D. Wheeler, D. Josell and T. P. Moffat, J. Electrochem. Soc., 2003, 150, C61–C66 CrossRef CAS.
- G. Baltrūnas, Electrochim. Acta, 2003, 48, 3659–3664 CrossRef.
- B. Bozzini, L. D'Urzo, C. Mele and V. Romanello, J. Phys. Chem. C, 2008, 112, 6352–6358 CAS.
- S. A. Hossain and M. Saitou, J. Appl. Electrochem., 2008, 38, 1653–1657 CrossRef.
- R. Bomparola, S. Caporali, A. Lavacchi and U. Bardi, Surf. Coat. Technol., 2007, 201, 9485–9490 CrossRef CAS.
- Y. B. Patil and K. M. Paknikar, Lett. Appl. Microbiol., 2000, 30, 33–37 CrossRef CAS.
- C. L. Lasko and M. P. Hurst, Environ. Sci. Technol., 1999, 33, 3622–3626 CrossRef CAS.
- D. G. Foster, Y. Shapir and J. Jorne, J. Electrochem. Soc., 2005, 152, C462–C465 CrossRef CAS.
- D. G. Foster, Y. Shapir and J. Jorné, J. Electrochem. Soc., 2003, 150, C375–C380 CrossRef CAS.
- D. Gonnissen, S. Vandeputte, A. Hubin and J. Vereecken, Electrochim. Acta, 1996, 41, 1051–1056 CrossRef CAS.
- S. Vandeputte, A. Hubin and J. Vereecken, Electrochim. Acta, 1997, 42, 3429–3441 CrossRef CAS.
- B.-G. Xie, J.-J. Sun, Z.-B. Lin and G.-N. Chen, J. Electrochem. Soc., 2009, 156, D79–D83 CrossRef CAS.
- M. Miranda-Hernández and I. González, J. Electrochem. Soc., 2004, 151, C220–C228 CrossRef.
- B. J. Polk, M. Bernard, J. J. Kasianowicz, M. Misakian and M. Gaitan, J. Electrochem. Soc., 2004, 151, C559–C566 CrossRef CAS.
- J.-C. Bian, Z. Li, Z.-D. Chen, H.-Y. He, X.-W. Zhang, X. Li and G.-R. Han, Appl. Surf. Sci., 2011, 258, 1831–1835 CrossRef CAS.
- G. M. Oliveira, M. R. Silva and I. A. Carlos, J. Mater. Sci., 2007, 42, 10164–10172 CrossRef.
- Z.-B. Lin, B.-G. Xie, J.-S. Chen, J.-J. Sun and G.-N. Chen, J. Electroanal. Chem., 2009, 633, 207–211 CrossRef CAS.
- Z.-B. Lin, J.-H. Tian, B.-G. Xie, Y.-A. Tang, J.-J. Sun, G.-N. Chen, B. Ren, B.-W. Mao and Z.-Q. Tian, J. Phys. Chem. C, 2009, 113, 9224–9229 CAS.
- M.-C. Tsai, D.-X. Zhuang and P.-Y. Chen, Electrochim. Acta, 2010, 55, 1019–1027 CrossRef CAS.
- A. Liu, X. Ren, M. An, J. Zhang, P. Yang, B. Wang, Y. Zhu and C. Wang, Sci. Rep., 2014, 4, 3837 Search PubMed.
- A. Liu, X. Ren, B. Wang, J. Zhang, P. Yang, J. Zhang and M. An, RSC Adv., 2014, 4, 40930–40940 RSC.
- M. Puszyńska-Tuszkanow, T. Grabowski, M. Daszkiewicz, J. Wietrzyk, B. Filip, G. Maciejewska and M. Cieślak-Golonka, J. Inorg. Biochem., 2011, 105, 17–22 CrossRef.
- A. Liu, X. Ren, J. Zhang, G. Yuan, P. Yang, J. Zhang and M. An, New J. Chem., 2015, 39, 2409–2412 RSC.
- A. P. Abbott, M. Azam, G. Frisch, J. Hartley, K. S. Ryder and S. Saleem, Phys. Chem. Chem. Phys., 2013, 15, 17314–17323 RSC.
- G. M. de Oliveira, L. L. Barbosa, R. L. Broggi and I. A. Carlos, J. Electroanal. Chem., 2005, 578, 151–158 CrossRef CAS.
- G. M. de Oliveira, M. R. Silva and I. Carlos, J. Mater. Sci., 2007, 42, 10164–10172 CrossRef CAS.
- J. C. Ballesteros, E. Chaînet, P. Ozil, G. Trejo and Y. Meas, J. Electroanal. Chem., 2010, 645, 94–102 CrossRef CAS.
- F. Endres and W. Freyland, J. Phys. Chem. B, 1998, 102, 10229–10233 CrossRef CAS.
- J. V. Zoval, R. M. Stiger, P. R. Biernacki and R. M. Penner, J. Phys. Chem., 1996, 100, 837–844 CrossRef CAS.
- N. Hernández, J. M. Ortega, M. Choy and R. Ortiz, J. Electroanal. Chem., 2001, 515, 123–128 CrossRef.
- J. M. Ortega, Thin Solid Films, 2000, 360, 159–165 CrossRef CAS.
- J. P. Franey, G. W. Kammlott and T. E. Graedel, Corros. Sci., 1985, 25, 133–143 CrossRef CAS.
- C. Kleber, R. Wiesinger, J. Schnöller, U. Hilfrich, H. Hutter and M. Schreiner, Corros. Sci., 2008, 50, 1112–1121 CrossRef CAS.
- L. Paussa, L. Guzman, E. Marin, N. Isomaki and L. Fedrizzi, Surf. Coat. Technol., 2011, 206, 976–980 CrossRef CAS.
- S. Z. El Abedin, E. M. Moustafa, R. Hempelmann, H. Natter and F. Endres, Electrochem. Commun., 2005, 7, 1111–1116 CrossRef.
- A. E. Alvarez and D. R. Salinas, J. Electroanal. Chem., 2004, 566, 393–400 CrossRef CAS.
- Y. Xiaowei, A. Maozhong, Z. Yunwang and Z. Lin, Electrochim. Acta, 2011, 58, 516–522 CrossRef.
- X. Ren, Y. Song, A. Liu, J. Zhang, P. Yang, J. Zhang, G. Yuan, M. An, H. Osgood and G. Wu, RSC Adv., 2015, 5, 64806–64813 RSC.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2000 Search PubMed.
- B. Scharifker and G. Hills, Electrochim. Acta, 1983, 28, 879–889 CrossRef CAS.
- M. E. Hyde and R. G. Compton, J. Electroanal. Chem., 2003, 549, 1–12 CrossRef CAS.
- A. I. Bhatt and A. M. Bond, J. Electroanal. Chem., 2008, 619–620, 1–10 CrossRef CAS.
- P. He, H. Liu, Z. Li, Y. Liu, X. Xu and J. Li, Langmuir, 2004, 20, 10260–10267 CrossRef CAS.
- M. Miranda-Hernández, M. Palomar-Pardavé, N. Batina and I. González, J. Electroanal. Chem., 1998, 443, 81–93 CrossRef.
- D. Gonnissen, W. Simons and A. Hubin, J. Electroanal. Chem., 1997, 435, 149–155 CrossRef CAS.
- Z. Zhang and D. P. Barkey, J. Electrochem. Soc., 2007, 154, D550–D556 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |