
A ratiometric, fluorescent BODIPY-based probe for transition and heavy metal ions†
Abstract
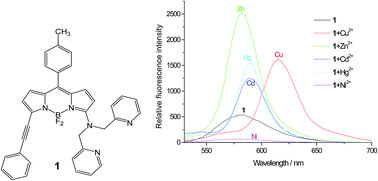
A novel metal ion-sensitive fluorescent probe – 4,4-difluoro-8-(4-methylphenyl)-5-(phenylethynyl)-3-[bis(pyridin-2-ylmethyl)amino]-4-bora-3a,4a-diaza-s-indacene – based on the BODIPY platform with di(2-picolyl)amine as chelator has been synthesized and spectroscopically and photophysically characterized. The generalized treatment of the solvent effect shows that solvent dipolarity is primarily responsible for the observed shifts of the absorption and fluorescence emission maxima. Complex formation with various metal ions is investigated in acetonitrile solution by means of spectrophotometric and fluorometric titrations. The BODIPY indicator forms 1 : 1 complexes with several transition metal (Ni2+, Cu2+, Zn2+) and heavy metal (Cd2+, Hg2+) ions, producing large bathochromic shifts in the absorption and fluorescence spectra and, except for Ni2+, cation-induced fluorescence amplifications. The dissociation constants of the metal ion complexes range from 4 μM for Hg2+ to 48 μM for Zn2+.
- This article is part of the themed collection: Luminescence and photophysical properties of metal complexes
 Please wait while we load your content...
Please wait while we load your content...

