
Selective and comprehensive characterization of the quinochalcone C-glycoside homologs in Carthamus tinctorius L. by offline comprehensive two-dimensional liquid chromatography/LTQ-Orbitrap MS coupled with versatile data mining strategies†
Abstract
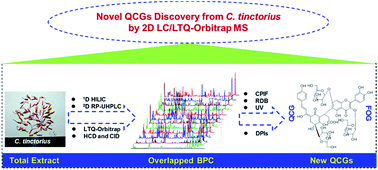
Quinochalcone C-glycosides (QCGs) are a series of pharmacologically bioactive components chemotaxonomic for Carthamus tinctorius L. The low abundance and ubiquitous interference from flavonoid O-glycosides (FOGs) frequently hinder the systematic exposure and characterization of these QCG homologs. We herein present an offline comprehensive two-dimensional liquid chromatography/linear ion-trap quadrupole/Orbitrap mass spectrometry (2D LC/LTQ-Orbitrap MS) approach coupled with versatile data mining strategies, to systematically characterize the QCGs from C. tinctorius. Initially, an offline 2D LC system, with an orthogonality of 71% and a theoretical peak capacity of 7654, was established by combining an Acchrom XAmide column and a BEH Shield RP-18 column. Subsequently, the water extract of C. tinctorius was separated by first dimensional hydrophilic interaction liquid chromatography (HILIC) yielding twelve fractions, which were further analyzed by reversed-phase ultra-high performance liquid chromatography/LTQ-Orbitrap MS using high energy C-trap dissociation (HCD) and collision-induced dissociation (CID) in the negative ion mode. The characteristic product ion filtering of m/z 119.05 (C8H7O−) in the HCD spectra, ring double bond equivalent (RDB 10–30), characteristic UV absorption around 405 nm, preferred 0,2X0 cleavage for C-glycosides, and diagnostic product ions analysis, were simultaneously employed for the structural elucidation of QCGs. Ultimately, 163 QCQ homologs were putatively characterized, and 149 are potential new ones. Particularly, nine dimers of QCG-FOG have not been previously reported. The obtained results have greatly expanded the knowledge on the structural diversity of QCGs, demonstrating the potency of the offline comprehensive 2D LC/LTQ-Orbitrap MS approach in separation and characterization of minor herbal components.
 Please wait while we load your content...
Please wait while we load your content...

