
Designing binder-free, flexible electrodes for high-performance supercapacitors based on pristine carbon nano-onions and their composite with CuO nanoparticles†
Abstract
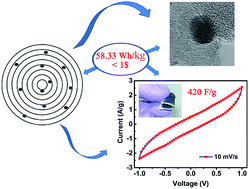
The increasing demand for energy has triggered tremendous research efforts for the development of light-weight and durable energy storage devices. This requires exploring simple and economical methods to prepare the active materials and to design lightweight, flexible, free-standing supercapacitor electrodes in an inexpensive binder-free process. Herein, we try to address both these critical issues using CNOs and their composite with CuO as the active material. Active materials were supported on cotton wipes by a simple “sonication and drying” process to obtain light-weight, flexible and free-standing binder-free electrodes. In a symmetrical two-electrode cell, a pristine CNO electrode delivers a specific capacitance of 102.16 F g−1 (20 mV s−1), an energy density of 14.18 W h kg−1 and a power density of 2448 W kg−1, which are the highest values reported so far for CNO-based materials. CNO–CuO nanocomposites demonstrate a very significant specific capacitance of 420 F g−1 (10 mV s−1) with deliverable energy and power density at 58.33 W h kg−1 and 4228 W kg−1, respectively. Electrodes of both the active materials show an excellent cyclic performance and stability, retaining up to 90–95% of their initial capacitance after 5000 charge–discharge cycles at a current density of 5 A g−1. A simple cost estimation indicates that our device can deliver an energy density of 58.33 W h kg−1 at an estimated cost of less than 1 $.
 Please wait while we load your content...
Please wait while we load your content...

