
Hydrogen-treated BiFeO3 nanoparticles with enhanced photoelectrochemical performance
Chao Zhanga,
Yanhong Lia,
Mengsha Chua,
Nannan Rongb,
Peng Xiao*a and
Yunhuai Zhang*b
aCollege of Physics, Chongqing University, Shapingba, Chongqing, 401331, China. E-mail: xiaopeng@cqu.edu.cn; xp2031@163.com; Fax: +86-023-65102031; Tel: +86-15823038874
bCollege of Chemistry and Chemical Engineering, Chongqing University, Shapingba, Chongqing, 400044, China
First published on 1st March 2016
Abstract
Enhancing charge separation and collection in semiconducting photoelectrodes is a key issue in the area of photoelectrochemical applications. Herein, we report that the photoelectrochemical performance of BiFeO3 nanoparticles can be significantly enhanced by treatment with hydrogen atmosphere at elevated temperature for different time periods. Compared with pristine BiFeO3 nanoparticles, the hydrogen-treated BiFeO3 nanoparticles showed substantial improvement of intrinsic features from structural, optical and electronic aspects. These hydrogen-treated BiFeO3 nanoparticles exhibit an unexpected red shift, which was attributed to the formation of sub-band-gap states between the conduction and valence bands, resulting from oxygen vacancies due to the hydrogen treatment. However, the highly concentrated defects in BiFeO3 nanoparticles can act as charge annihilation centers, which is not conducive to the separation of photo-generated carriers. The hydrogen-treated BiFeO3 prepared at 300 °C for 15 min exhibited optimal photoelectrochemical performance. More than three times higher photocurrent density was achieved compared with pristine BiFeO3, demonstrating its potential in practical application.
1. Introduction
Photoelectrochemical water splitting into hydrogen and oxygen is a promising and environmentally friendly method to harvest solar energy.1–5 In comparison to the photovoltaic approach, this method converts the solar energy into chemical energy, which is much easier to store than electricity.6 Multiferroic materials have recently been used in both photovoltaic and photoelectrochemical water splitting due to their ferroelectric properties and relatively narrow band gaps, which can help to enhance the separation efficiency of the photo-generated charges and allow them to harness the majority of solar radiation in the visible range, respectively.7–11 As a typical perovskite type of multiferroic material, bismuth ferrite (BiFeO3–BFO), with simultaneous ferroelectric and ferromagnetic ordering, has recently been recognized as a potential system for photoelectrochemical applications because of its narrow band gap (Eg = 2.2–2.7 eV) and good chemical stability.7–9 And, it has been regarded as one of the third-generation photocatalysts.12 However, the investigations of BFO nanomaterials for photoelectrochemical water splitting are still limited because of the quick recombination of photo-generated charge carriers.In recent years, many researchers have been working to promote the photoactivity of BFO materials, and some strategies such as material structural designing,9,10,13–15 noble metal decorating,14–16 elemental doping,17,18 and heterojunction19–22 have been reported. For instance, the construction of BFO-based heterostructured composites with suitable banding alignment can greatly facilitate the separation of photo-generated electron–hole pairs and suppress their recombination. In this respect, plentiful BFO-based heterojunction photocatalysts such as BiFeO3/TiO2,19,20 BiFeO3–graphene,21,22 have been widely developed and exhibited enhanced photoactivity. In addition, decorating noble metals such as Au14 and Ag15,16 have been proven to significantly improve the photoactivity of BFO due to the Schottky junction and localized surface plasmon resonance (LSPR). However, the high cost of noble metals severely hinders their practical application. Importantly, these strategies mentioned above do not change and/or improve the intrinsic properties of BFO such as optical absorption property and charge transfer ability. So it is highly desirable to intrinsically improve the photoactivity of the BFO with an environmentally friendly and cost-effective method.
Recently, Wang et al. demonstrated that photoactivity of hydrogen-treated TiO2 nanowires can be enhanced as a result of increased donor density and electrical conductivity by introducing a moderate amount of oxygen vacancies.23 It is well known that oxygen vacancy is a fundamental and intrinsic defect in metal oxide semiconductors and has a critical effect on their electronic and physicochemical properties such as electronic band structures and optical absorption.24–26 Moreover, these oxygen vacancies could act as electron scavengers delaying electron–hole pair recombination during the photoreaction process, thus improving the separation efficiency of photo-generated electrons and holes.24 Therefore, in this study, oxygen-deficient BFO nanoparticles were readily obtained by sintering in hydrogen atmosphere and we demonstrated for the first time that the light-harvesting ability and the separation efficiency of the photo-generated electrons and holes of BFO nanoparticles can be improved simultaneously and significantly through creating oxygen vacancies within BFO. The optimized BFO photoelectrode showed a remarkable photocurrent of 0.69 μA cm−2 at 0.8 V vs. Ag/AgCl, which was over 3 times higher than that without the hydrogen treatment. The hydrogen-treated BFO (H-BFO) with high efficiency could also be used as a potential material to achieve better performance for photovoltaic applications.
2. Experimental section
2.1 Materials and synthesis
The chemicals used in the synthesis were bismuth nitrate pentahydrate (Bi(NO3)3·5H2O, Aladdin, 99%), iron nitrate nonahydrate (Fe(NO3)3·9H2O, Aladdin, 98.5%), and N,N-dimethylfformamide (DMF, C3H7NO, 99.5%, ChengDu KeLong Chemical Co. Ltd.). All the reagents were of analytical grade and used without any purification.A typical preparation process used to obtain the precursors for the synthesis of BiFeO3 nanoparticles is as follows: Bi(NO3)3·5H2O and Fe(NO3)3·9H2O with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio were successively added to the DMF solvent. The resulting mixture was stirred vigorously with a magnetic stirrer to obtain homogeneous precursor solution at room temperature. The resulted precursor solution was heated at 100 °C on a hot plate for evaporating of solvent until forming the gel. Then the gel was preheated at 350 °C for 60 min to eliminate the organic components and finally the samples were calcined at 550 °C for 120 min in ambient air for crystallization. After cooling down to room temperature naturally, the remains were ground for 2 h. Then the as prepared BiFeO3 nanoparticles were obtained.
:1 molar ratio were successively added to the DMF solvent. The resulting mixture was stirred vigorously with a magnetic stirrer to obtain homogeneous precursor solution at room temperature. The resulted precursor solution was heated at 100 °C on a hot plate for evaporating of solvent until forming the gel. Then the gel was preheated at 350 °C for 60 min to eliminate the organic components and finally the samples were calcined at 550 °C for 120 min in ambient air for crystallization. After cooling down to room temperature naturally, the remains were ground for 2 h. Then the as prepared BiFeO3 nanoparticles were obtained.
A drop casting method was used to fabricate the BFO film. In the typical procedure, 4 mg BFO nanoparticles were dispersed in 1 mL absolute ethylalcohol, followed by ultrasonically treating the suspension for 2 h. The suspension was dropped cast onto a Sn-doped In2O3 (ITO, 85% transmittance in the visible, 15 Ω cm−2, purchased from Zhuhai Kaivo Electronic Components Co. Ltd.) substrate. The drop casting process was repeated several times to achieve a BFO film with mass loading of about 0.128 mg cm−2.
In order to prepare slightly hydrogen-treated BFO, the above BFO samples were transferred into a tube furnace system for hydrogen treatment, where the samples were heated in a mixed gas atmosphere of 95% Ar and 5% H2 at 300 °C for 5 min, 10 min, 15 min, 30 min and 60 min, respectively. The gas flow rate in the furnace was about 50 sccm. After this treatment, the H-BFO samples were obtained and denoted as H-BFO-5, H-BFO-10, H-BFO-15, H-BFO-30 and H-BFO-60, respectively.
2.2 Material characterization
Powder X-ray diffraction analysis was measured by using a BrukerD8 Advance X-ray diffractometer operating at 40 kV and 40 mA using Cu Kα radiation. The morphologies of the samples were characterized using field emission scanning electron microscopy (FESEM, FEI Nova 400 Nano-SEM). UV-Vis absorption spectra were recorded using a UV-visible (UV3600, Shimadzu) spectrophotometer with BaSO4 as the background between 200 and 800 nm. Raman spectroscopy was performed at the excitation with an argon ion laser of 532 nm (Horiba Lab). Steady state photoluminescence (PL) of samples dispersed in ethanol (0.5 mg mL−1) was measured using an excitation wavelength of 387 nm on a RF-5301pc spectrofluorometer (Shimadzu Corporation). The surface composition of the sample was evaluated by X-ray photoelectron spectra (XPS, ESCALAB 250 Thermo Fisher Scientific). All the measurements were performed at room temperature.2.3 Photoelectrochemical measurement
The photoelectrochemical measurements were performed in a photoelectrochemical cell with a quartz window for illumination. The photocurrents were measured by a CHI 660D electrochemical workstation in a standard three electrode system using the as-prepared film as the working electrode with an active area of ca. 1 cm2, a platinum foil (2 × 4 cm2) as counter electrode and saturated Ag/AgCl as reference electrode. 0.2 M sodium sulphate (Na2SO4) aqueous solution was used as the electrolyte (pH = 6.3). The sunlight was simulated by a 300 W Xenon lamp (Spectra Physics). The scan rate was 20 mV s−1. Mott–Schottky measurements were measured in 0.2 M Na2SO4 solution in the dark at a frequency of 1 kHz and the scan rate was 50 mV s−1.2.4 Photocatalytic activity test
The photocatalytic activities of the as-prepared samples were evaluated by the degradation of Rhodamine-B (RhB) in aqueous solution using a 300 W Xenon lamp. The initial concentration of RhB was 10 mg L−1, and in a typical experiment, 100 mL of the RhB solution was continuously stirred and 50 mg of the as-prepared BFO powder was putted into the RhB solution (in dark and waiting for 60 min) to obtain a good dispersion and adsorption equilibrium. Then the mixed solution was illuminated by a Xenon lamp in the photochemical reactor. In order to monitor the RhB concentration in the solution, 5 mL of the solution was taken from the photochemical reactor at an interval of 60 min and centrifugated. The concentration of the RhB solution was determined by the intensity of absorption peak of RhB at 553 nm using a UV-vis spectrophotometer, from which the percentage of degradation was determined.3. Results and discussion
3.1 Structural and compositional characterization
XRD patterns were collected from the pristine BFO and H-BFO with different hydrogenation time to check the crystalline properties and the results are shown in Fig. 1. It could be identified that all the diffraction peaks can be indexed to distorted rhombohedral perovskite structure with the space group of R3c for BFO (JCPDS file no. 86-1518). No traces of other phases such as Bi2Fe4O9 and Bi2O3/Fe2O3 were detected in the XRD patterns, revealing high purity of the as-prepared products. The average grain size of the sample can be evaluated from (101), (012), and (110) peaks according to Scherer equation: τ = (kλ)/(βcosθ), where τ is the mean size of the ordered (crystalline) domains, which may be smaller than or equal to grain size, k is the shape factor with a typical value of 0.9, λ is the X-ray wavelength, β is the line broadening full width at half maximum (FWHM) peak height in radians, and θ is the Bragg angle.27 The calculated average crystalline grain size of pristine BFO, H-BFO-15, H-BFO-30, and H-BFO-60 were 20.2 nm, 18.0 nm, 17.9 nm and 16.7 nm, respectively. Obviously, the average sizes of the crystalline grains of H-BFO were slightly smaller than pristine BFO. These results suggested that some disordered phases were likely formed on the surface of the crystalline phase of BFO nanoparticles after hydrogenation, which has also been observed in other studies.28,29 Furthermore, some peaks tended to merge and the peak intensities decreased with increasing the hydrogen treatment time. In particular, the peaks corresponding to (104) and (110) planes at 2θ values of 32° merging into a single peak and significant reduction in diffraction intensities could be clearly identified for the H-BFO-60 sample. These results suggested gradual degradation of BFO crystallinity, which was expected for the increased amount of defects. It has also been observed in the recent studies about hydrogenated TiO2.23,29
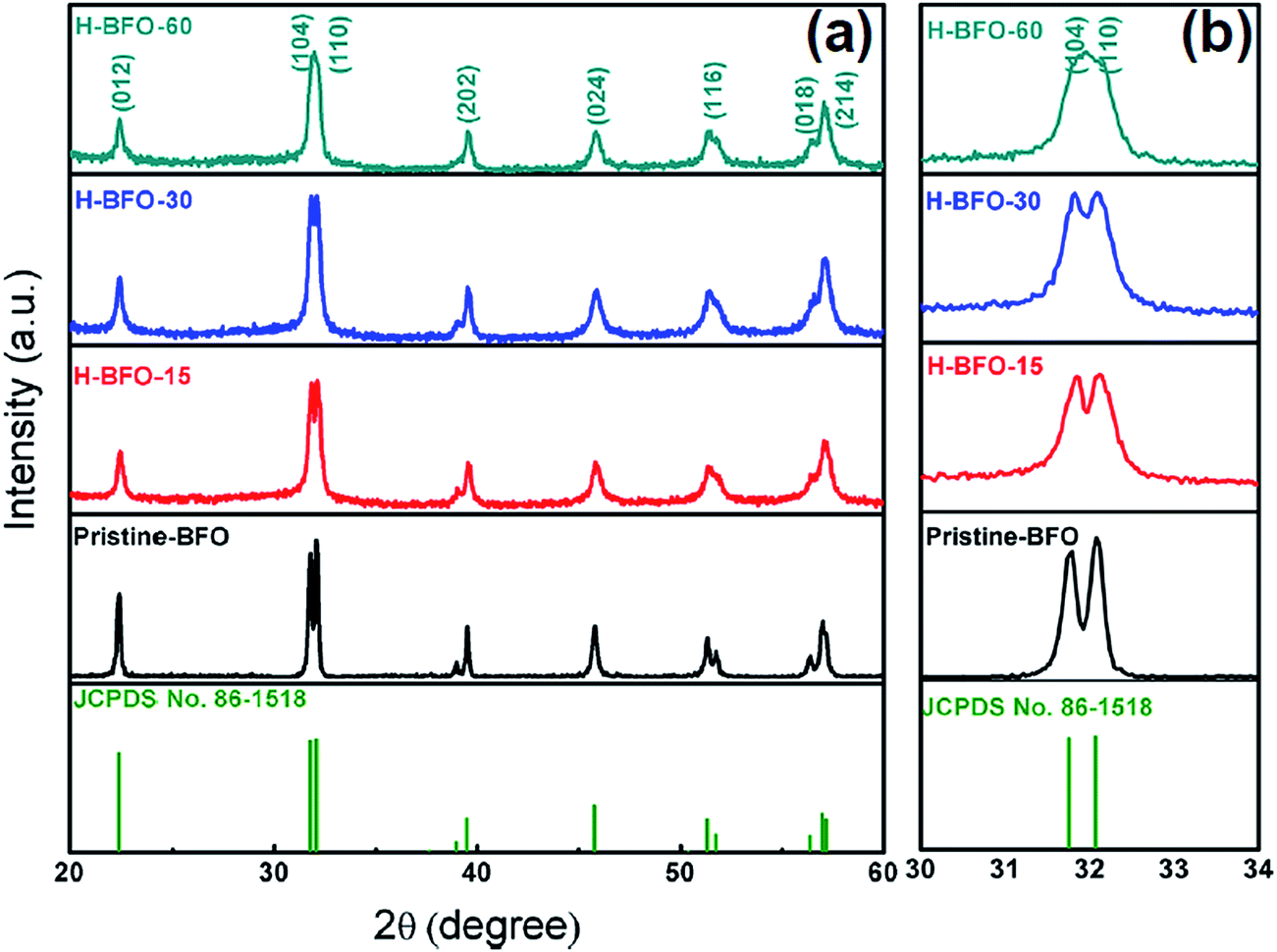 | ||
| Fig. 1 (a) XRD patterns of pristine BFO, H-BFO-15, H-BFO-30, and H-BFO-60 samples. (b) Enlarge view of diffraction peaks around 2θ = 32°. | ||
The SEM images of the pristine BFO and H-BFO are shown in Fig. 2. As we can see, the pristine BFO exhibits a largely spherical morphology that consists of loosely bound agglomerates. The formation of agglomerated clusters indicated that the sample was partly melted under high temperature.30 Additionally; the morphologies of hydrogen treated samples almost remain unchanged as shown in Fig. 2b–d, which suggests that the hydrogen annealing treatment did not change the morphology of the as-prepared BFO sample.
 | ||
| Fig. 2 SEM images of (a) pristine BFO, (b) H-BFO-15, (c) H-BFO-30, and (d) H-BFO-60. | ||
To get further insight on the structural change of the BFO samples with different hydrogenation degree, typical Raman spectra tests were performed within wave number range 50–700 cm−1 as shown in Fig. 3. The sharp Raman peaks implies that the BFO nanoparticles were well crystalline. At room temperature, BFO presents distorted rhombohedral perovskite structure with the space group of R3c, hence 13 (4A1 + 9E) Raman modes are expected according to the group theory.31,32 Pristine BFO showed distinct and sharp Raman peaks at around 136 cm−1, 170 cm−1, 217 cm−1, and a week intensity peak at 475 cm−1, which can be assigned to A1-1, A1-2, A1-3 and A1-4, respectively. The peaks around 75 cm−1, 260 cm−1, 345 cm−1, 365 cm−1, 469 cm−1, and 520 cm−1 are assigned to E1, E2, E3, E4, E5, and E6, respectively. The above result is in good agreement with previous reports.31,32 As the hydrogen treatment time increased, the Raman peaks become broader, and the peak intensity gradually decreased, suggesting gradual degradation of BFO crystallinity, which most likely arises from the formation of defects, such as oxygen vacancies and hydrogen impurities.33–35 In addition, we also found that the color of the BFO nanoparticles was changed after the hydrogen treatment. As shown in Fig. 4a, pristine BFO shows red rust in color. The colors of H-BFO gradually become darker as increasing the time of hydrogen treatment. The variation in color suggests that the band gap of BFO may be changed as a result of hydrogen treatment. To confirm this prediction, the UV-Vis absorption spectra of pristine BFO and H-BFO were collected.
 | ||
| Fig. 3 Raman spectra of pristine BFO, H-BFO-15, H-BFO-30, and H-BFO-60 measured under an excitation of a 532 nm Ar+ laser. | ||
 | ||
| Fig. 4 (a) Related photographs and (b) UV-vis absorption spectra of the pristine–BFO, H-BFO-15, H-BFO-30, and H-BFO-60 samples. The insert is the calculation diagram of their band gaps. | ||
Shown in Fig. 4b are the UV-Vis absorption spectra of pristine BFO and hydrogen-treated BFO samples. It is apparently that the absorption of H-BFO in the region of visible light gradually rises to a high level with the extension of hydrogenation time. To estimate the band gaps, the absorption curves were replotted according to the following equation (αhν) = A(hν − Eg)n/2.12 In the equation, α is the absorption coefficient, hν is the photon energy, A is a constant relative to the material, Eg is the absorption band-gap energy, and n is 1 and 4 for a direct- and indirect-band gap semiconductor, respectively. Considering that BFO is a direct band gap material, the value of n for BFO is 1.36 The corresponding values of direct band gap of pristine BFO and H-BFO can be evaluated by extrapolating the linear portion of curve to x axis. Thus the band gaps were estimated to be 2.10 eV, 2.00 eV, 1.97 eV, and 1.87 eV for pristine BFO, H-BFO-15, H-BFO-30, and H-BFO-60, respectively. The direct band gap values were consistent with those reported for BFO nanostructures (1.8–2.5 eV),10,37 which are smaller than that of bulks or thin films (2.66–2.81 eV).36,38,39 One can see that the band gap gradually narrowed with the extension of hydrogen treatment time, which is agreement with the color changes from red rust to dark. This may be due to the creation of oxygen vacancies by hydrogen treatment, leading to sub-band-gap transition corresponding to the excitation from the valence band to the impurity band. Accordingly, the band-to-band transition as well as the sub-band-gap transition concurrently happened in the hydrogen treatment samples, where band-to-band transition only occurred in the pristine BFO.
In order to evaluate the efficiency of charge trapping and recombination of photo-induced electrons and holes in the BFO after hydrogenation, the photoluminescence (PL) emission was measured as well. Fig. 5 shows the PL spectra of BFO with and without hydrogen treatment in the wavelength range of 450–600 nm using 387 nm as excitation wavelength. As we can see, two emission peaks were detected at around 485 and 530 nm corresponded to the energy levels of 2.55 and 2.33 eV, respectively, which consistent with those reported in the literature.40 By contrast, the hydrogen treated samples exhibit a lower emission intensity than pristine BFO, indicating that the recombination rate of electrons and holes is inhibited considerably in hydrogen-treated BFO.
 | ||
| Fig. 5 Photoluminescence spectra of pristine BFO, H-BFO-15, H-BFO-30, and H-BFO-60 measured under an excitation of 387 nm Ar+ laser. | ||
To shed light on the mechanism of hydrogen treatment, X-ray photoelectron spectroscopy (XPS) was employed to elucidate the electronic structure and surface state of hydrogen treated BFO nanostructures. As shown in Fig. 6a, the XPS survey spectra collected from pristine BFO and H-BFO-15 sample are very similar. Besides weak carbon signals (which were believed to be induced during sample preparation and subsequent handing), only Bi, Fe and O core levels were observed, indicating that hydrogen treatment is a clean process that does not introduce impurities into the BFO nanostructures. Fig. 6b–d show the high resolution Bi 4f, Fe 2p and O 1s spectra of the pristine BFO and H-BFO-15 samples. The Bi 4f XPS spectra are identical with Bi 4f7/2 and Bi 4f5/2 peaks centered at binding energies of 158.45 and 163.78 eV, which are consistent with the typical values for BFO.41 Fig. 6c compares the core level Fe 2p XPS spectra of pristine BFO and H-BFO-15 samples. The peaks with binding energy of 710.47 and 723.49 eV are the characteristic Fe 2p3/2 and Fe 2p1/2 peaks of BFO, respectively. A weak satellite peak is also observed nearly 8 eV above the 2p3/2 principal peak for pristine BFO, which can be attributed to the characteristic of the oxidation state of the Fe3+.42 But for hydrogen-treated BFO, the small satellite peak was substituted by a shoulder peak, which may indicate the partial presence of Fe2+ states, probably resulting from the emergence of oxygen vacancies in the intrinsic material. The O 1s XPS spectrum of the BFO at binding energy 530.4 eV is consistent with the typical value reported for BFO;41 while, the hydrogen-treated BFO exhibits a broader O 1s peak with an additional shoulder at higher binding energy. This broad peak can be deconvoluted into two peaks centered at 529.44 and 531.27 eV.43,44 The peak at about 529.44 eV is due to oxygen in the crystal lattice and the peak at about 531.27 eV can be attributed to surface hydroxyl group.20,41,43 In general, hydroxyl groups are bonded to the metal cations to compensate the charge balance in the oxygen deficient surface region. Thus, the appearance of hydroxyl group indicates the formation of oxygen vacancies after hydrogen treatment.
 | ||
| Fig. 6 XPS spectra of the pristine BFO and H-BFO-15 sample. (a) Survey XPS spectra; (b–d) high-resolution XPS core spectra of Bi 4f, Fe 2p, and O 1s, respectively. | ||
3.2 Photoelectrochemical studies
The photoelectrochemical properties of pristine BFO and hydrogen-treated BFO samples were measured in a standard three-electrode electrochemical cell (using BFO films as working electrode, a platinum as the counter electrode, and Ag/AgCl in saturated KCl as the reference) with 0.2 M Na2SO4 solution as electrolyte (Experimental section). Fig. 7a shows the linear sweep voltammograms for pristine BFO and hydrogen-tread BFO samples. For the H-BFO samples, the photocurrent densities significantly increase. Notably, compared with pristine BFO, a negative shift was observed in the onset potential. This was also elucidated by the formation of sub-band-gap states after hydrogen treatment, which shifted the onset potential toward the negative potential. As shown in Fig. 7b, the transient photocurrent responses are presented. We observe that the photocurrent increases rapidly upon xenon solar light irradiation and finally reaches saturation which is called the ‘on’ state. The photocurrent decreases quickly in a short time and recovers to the original dark current value called the ‘off’ state. As increasing the hydrogen treatment time, the photocurrent densities of H-BFO firstly present drastic increase, then, decrease. After five on–off cycles, the current densities of the pristine BFO, H-BFO-5, H-BFO-10, H-BFO-15, H-BFO-30 and H-BFO-60 electrodes were 0.12, 0.17, 0.48, 0.69, 0.44, and 0.32 μA cm−2, respectively, as shown in Fig. 7c. Obviously, the H-BFO-15 sample shows the highest photocurrent density, which is about 3.2 times higher than that of pristine BFO electrode. The obvious enhancement of hydrogen-treated samples in photocurrent indicated smaller recombination and more efficient separation of photo-generated electron–hole pairs at their interface. The same experiments were repeated for several times and same results were obtained. Thus, there is reason to believe that hydrogen treatment could significantly enhance the photoelectrochemical performance of BFO. The enhancement is ascribed to the increased amount of oxygen vacancies which can significantly improve the photocurrent. However excess vacancies would result in a recombination of photo-generated holes with electrons. Therefore, the optimized oxygen vacancies will not only improve the conductivity of BFO but also may reduce the recombination of photo-excited holes with electrons. | ||
| Fig. 7 (a) Linear sweeps voltammograms and (b) photocurrents responses of pristine BFO, H-BFO-5, H-BFO-10, H-BFO-15, H-BFO-30, and H-BFO-60 samples. (c) Photocurrents values of pristine BFO, H-BFO-5, H-BFO-10, H-BFO-15, H-BFO-30, and H-BFO-60 samples after five on–off cycles. | ||
For deep-seated insight into the effect of hydrogen treatment on the electronic properties of BFO, we measured the electrochemical impedance for pristine BFO and hydrogen-treated BFO nanoparticles. Fig. 8 shows the Mott–Schottky plots, which were generated from the capacitance values that were derived from the electrochemical impedance obtained at each potential at 1 kHz frequency. All samples show positive slopes in the Mott–Schottky, as expected for typical n-type semiconductors. According to the theory, the donor density of these n-type BFO samples can be calculated from the slope of Mott–Schottky plots using the following equation
| Nd = (2/e0εε0)[d(1/C2)/dV]−1 |
 | ||
| Fig. 8 Mott–Schottky plots collected at a frequency of 1 kHz in the dark for the pristine BFO, H-BFO-15, H-BFO-30, and H-BFO-60 samples. | ||
Considering the practical application, the photocatalytic activities of the hydrogen-treated samples were also evaluated by degradation of RhB in aqueous solution under irradiation of a 300 W Xenon lamp. Fig. 9 shows the photodegradation efficiencies of RhB as a function of irradiation time over different BFO samples. In the absence of the BFO, the self-degradation of RhB under light irradiation was insignificant. After 300 min of irradiation, about 18.2% of RhB was decomposed with pristine BFO, while 53.3%, 40.4%, 25.1% for H-BFO-15, H-BFO-30, and H-BFO-60, respectively. In contrast, hydrogen-treated samples exhibited better photocatalytic activities and the H-BFO-15 showed the highest photocatalytic activity. As further increasing hydrogen treatment time, the photocatalytic dye degradation rate decreased, which was consistent with the photoelectrochemical test result. Combined with photoelectrochemical analysis, it is demonstrated that hydrogen treatment can be used as a general strategy to fundamentally improve the photoactivity of BFO.
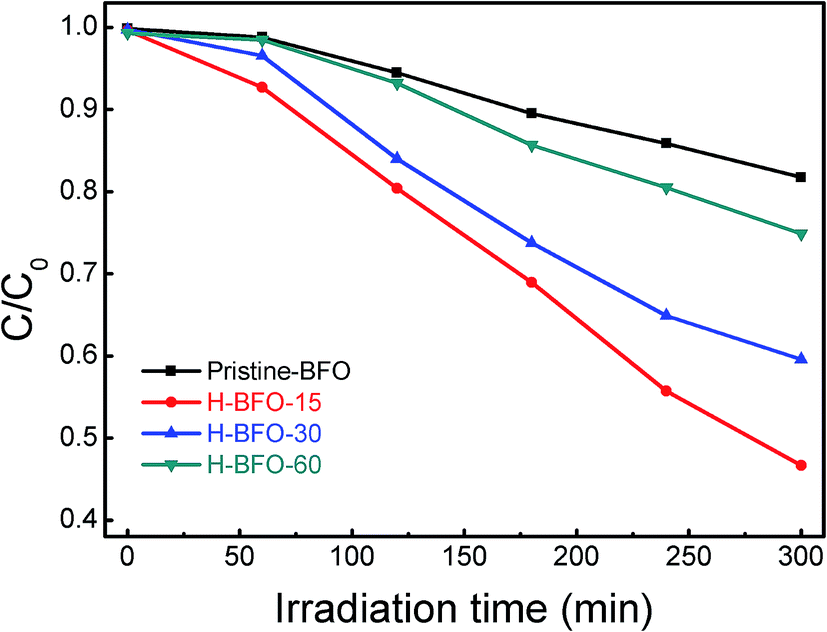 | ||
| Fig. 9 The photodegradation efficiencies of RhB solution as a function of irradiation time for the pristine BFO, H-BFO-15, H-BFO-30, and H-BFO-60 samples. | ||
4. Conclusions
In summary, hydrogen-treated BiFeO3 nanoparticles with different hydrogenation degrees are efficiently synthesized through annealing pristine BFO in hydrogen atmosphere for different time. Compared with pristine BFO, the hydrogen-treated BFO at 300 °C for 15 min exhibits enhanced photoelectrochemical performance. Such enhanced photoelectrochemical performance could be ascribed to the narrowed band gap and increased donor density as a result of formation of oxygen vacancies; while the highly concentrated defects in BFO act as charge annihilation centers and most of the photo-generated carriers are consumed through significantly enhanced non-radiative recombination, which strongly inhibits the photoactivity of BFO. This work has demonstrated that hydrogen treatment can be used as a general strategy to fundamentally improve the performance of BiFeO3 material for photoelectrochemical applications.Acknowledgements
This work was supported by the sharing fund of Chongqing University's large-scale equipment and the Chongqing University postgraduates' innovation project (2015).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- J. W. Sun, D. K. Zhong and D. R. Gamelin, Energy Environ. Sci., 2010, 3, 1252 CAS.
- J. K. Kim, K. Shin, S. M. Cho, T. Lee and J. H. Park, Energy Environ. Sci., 2011, 4, 1465 CAS.
- Y. C. Ling, G. M. Wang, D. A. Wheeler, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 2119 CrossRef CAS PubMed.
- J. E. Yourey and B. M. Bartlett, J. Mater. Chem., 2011, 21, 7651 RSC.
- W. Ji, K. Yao, Y. F. Lim, Y. C. Liang and A. Suwardi, Appl. Phys. Lett., 2013, 103, 062901 CrossRef.
- T. Choi, S. Lee, Y. J. Choi, V. Kiryukhin and S. W. Cheong, Science, 2009, 324, 63 CrossRef CAS PubMed.
- D. W. Cao, Z. J. Wang, Nasori, L. Y. Wen, Y. Mi and Y. Lei, Angew. Chem., Int. Ed., 2014, 53, 11027 CrossRef CAS PubMed.
- F. Gao, Y. Yuan, K. F. Wang, X. Y. Chen, F. Chen, J. M. Liu and Z. F. Ren, Appl. Phys. Lett., 2006, 89, 102506 CrossRef.
- U. A. Joshi, J. S. Jang, P. H. Borse and J. S. Lee, Appl. Phys. Lett., 2008, 92, 242106 CrossRef.
- R. Nechache, C. Harnagea, S. Licoccia, E. Traversa, A. Ruediger, A. Pignolet and F. Rosei, Appl. Phys. Lett., 2011, 98, 202902 CrossRef.
- S. Li, Y. H. Lin, B. P. Zhang, Y. Wang and C. W. Nan, J. Phys. Chem. C, 2010, 114, 2903 CAS.
- Y. N. Huo, M. Miao, Y. Zhang, J. Zhu and H. X. Li, Chem. Commun., 2011, 47, 2089 RSC.
- S. Li, J. M. Zhang, M. G. Kibria, Z. Mi, M. Chaker, D. L. Ma, R. Nechache and F. Rosei, Chem. Commun., 2013, 49, 5856 RSC.
- S. Mohan, B. Subramanian and G. Sarveswaran, J. Mater. Chem. C, 2014, 2, 6835 RSC.
- A. M. Schultz, Y. L. Zhang, P. A. Salvador and G. S. Rohrer, ACS Appl. Mater. Interfaces, 2011, 3, 1562 CAS.
- H. C. Wang, Y. H. Lin, Y. N. Feng and Y. Shen, J. Electroceram., 2013, 31, 271 CrossRef CAS.
- R. Q. Guo, L. Fang, W. Dong, F. G. Zheng and M. R. Shen, J. Phys. Chem. C, 2010, 114, 21390 CAS.
- A. S. Zhu, Q. D. Zhao, X. Y. Li and Y. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 671 CAS.
- Y. C. Yang, Y. Liu, J. H. Wei, C. X. Pan, R. Xiong and J. Shi, RSC Adv., 2014, 4, 31941 RSC.
- Z. X. Li, Y. Shen, Y. H. Guan, Y. H. Hu, Y. H. Lin and C. W. Nan, J. Mater. Chem. A, 2014, 2, 1967 CAS.
- Z. X. Li, Y. Shen, C. Yang, Y. C. Lei, Y. H. Guan, Y. H. Lin, D. B. Liu and C. W. Nan, J. Mater. Chem. A, 2013, 1, 823 CAS.
- G. M. Wang, H. Y. Wang, Y. C. Ling, Y. C. Tang, X. Y. Yang, R. C. Fitzmorris, C. C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026 CrossRef CAS PubMed.
- X. C. Zhang, L. J. Zhao, C. M. Fan, Z. H. Liang and P. Han, Comput. Mater. Sci., 2012, 61, 180 CrossRef CAS.
- X. H. Lu, G. M. Wang, S. L. Xie, J. Y. Shi, W. Li, Y. X. Tong and Y. Li, Chem. Commun., 2012, 48, 7717 RSC.
- G. M. Wang, Y. C. Ling, X. H. Lu, F. Qian, Y. X. Tong, J. Z. Zhang, V. Lordi, C. R. Leao and Y. Li, J. Phys. Chem. C, 2013, 117, 10957 CAS.
- T. Xia, N. Li, Y. L. Zhang, M. B. Kruger, J. Murowchick, A. Selloni and X. B. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 9883 CAS.
- T. Xia and X. B. Chen, J. Mater. Chem. A, 2013, 1, 2983 CAS.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746 CrossRef CAS PubMed.
- C. Hengky, X. Moya, N. D. Mathur and S. Dunn, RSC Adv., 2012, 2, 11843 RSC.
- M. K. Singh, H. M. Jang, S. Ryu and M. H. Jo, Appl. Phys. Lett., 2006, 88, 042907 CrossRef.
- N. V. Minh and D. V. Thang, J. Alloys Compd., 2010, 505, 619 CrossRef.
- G. M. Wang, Y. C. Ling, H. Y. Wang, X. Y. Yang, C. C. Wang, J. Z. Zhang and Y. Li, Energy Environ. Sci., 2012, 5, 6180 CAS.
- D. Wang, X. T. Zhang, P. P. Sun, S. Lu, L. L. Wang, C. H. Wang and Y. C. Liu, Electrochim. Acta, 2014, 130, 290 CrossRef CAS.
- Y. Yan, M. Y. Han, A. Konkin, T. Koppe, D. Wang, T. Andreu, G. Chen, U. Vetter, J. R. Morante and P. Schaaf, J. Mater. Chem. A, 2014, 2, 12708 CAS.
- J. F. Ihlefeld, N. J. Podraza, Z. K. Liu, R. C. Rai, X. Xu, T. Heeg, Y. B. Chen, J. Li, R. W. Collins, J. L. Musfeldt, X. Q. Pan, J. Schubert, R. Ramesh and D. G. Schlom, Appl. Phys. Lett., 2008, 92, 142908 CrossRef.
- M. Arora, P. C. Sati, S. Chauhan, S. Chhoker, A. K. Panwar and M. Kumar, J. Supercond. Novel Magn., 2012, 26, 443 CrossRef.
- V. Fruth, E. Tenea, M. Gartner, M. Anastasescu, D. Berger, R. Ramer and M. Zaharescu, J. Eur. Ceram. Soc., 2007, 27, 937 CrossRef CAS.
- A. Kumar, R. C. Rai, N. J. Podraza, S. Denev, M. Ramirez, Y. H. Chu, L. W. Martin, J. Ihlefeld, T. Heeg, J. Schubert, D. G. Schlom, J. Orenstein, R. Ramesh, R. W. Collins, J. L. Musfeldt and V. Gopalan, Appl. Phys. Lett., 2008, 92, 121915 CrossRef.
- K. Prashanthi, G. Thakur and T. Thundat, Surf. Sci., 2012, 606, L83 CrossRef CAS.
- S. Chatterjee, A. Bera and A. J. Pal, ACS Appl. Mater. Interfaces, 2014, 6, 20479 CAS.
- W. Eerenstein, F. D. Morrison, J. Dho, M. G. Blamire, J. F. Scott and N. D. Mathur, Science, 2005, 307, 1203 CrossRef CAS PubMed.
- L. Fang, J. Liu, S. Ju, F. G. Zheng, W. Dong and M. R. Shen, Appl. Phys. Lett., 2010, 97, 242501 CrossRef.
- R. Das, G. G. Khan, S. Varma, G. D. Mukherjee and K. Mandal, J. Phys. Chem. C, 2013, 117, 20209 CAS.
| This journal is © The Royal Society of Chemistry 2016 |