
Synthesis of heteroatom-carbon nanosheets by solution plasma processing using N-methyl-2-pyrrolidone as precursor†
Abstract
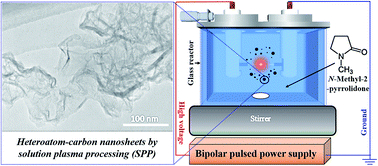
Nitrogen-carbon nanosheets (NCNS), composed of multi-layer graphene with turbostratic stacking, were successfully synthesized through a solution plasma processing (SPP) at room temperature and an atmospheric pressure. The plasma was generated in 200 mL of N-methyl-2-pyrrolidone (NMP), which was applied as the carbon and nitrogen precursors. The NCNS presented an electrical resistivity of 0.065 Ω cm, which is comparable with that of N-doped carbon nanofibers (CNFs) and N-doped carbon nanotubes (CNTs). The synthesis rate of NCNS was 20 mg min−1. From the characteristics analyses, NCNS showed the surface area of 277 m2 g−1, a pore volume of 0.95 cm3 g−1 and a moderate nitrogen content of 1.3 at%. The synthesized NCNS also exhibited catalytic activity towards oxygen reduction reaction (ORR). This unique synthesis method can be applicable to synthesize multiple types of heteroatom-carbon nanosheets.
 Please wait while we load your content...
Please wait while we load your content...

