
Phytosteroids and triterpenoids with potent cytotoxicities from the leaves of Chisocheton cumingianus†
Abstract
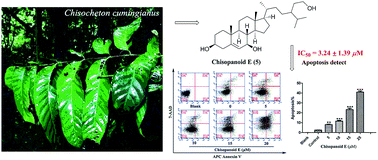
Six phytosteroids and three dammarane-type triterpenoids, namely chisopanoids A–F (1–6) and chisopanones G–I (7–9), together with nine known triterpenoids (10–18) were isolated from the leaves of Chisocheton cumingianus. Their structures were elucidated by extensive spectroscopic analysis, X-ray crystallographic analysis, Mosher's method and Mo2(OAc)4-induced electronic circular dichroism (ECD) data. All compounds were evaluated for their cytotoxicities against HepG2, U2OS and MCF-7 human cancer cell lines, as well as inhibitory effects on nitric oxide (NO) production in LPS-activated RAW 264.7 macrophages. Compounds 5 and 6 showed potent cytotoxicities towards MCF-7 with IC50 values of 3.24 ± 1.39 and 8.85 ± 4.73 μM, and were further proved to inhibit the cell proliferation mainly by inducing apoptosis.
 Please wait while we load your content...
Please wait while we load your content...

