
Energetic hybrid polymer network (EHPN) through facile sequential polyurethane curation based on the reactivity differences between glycidyl azide polymer and hydroxyl terminated polybutadiene†
Abbas
Tanver
a,
Fida
Rehman
a,
Aisha
Wazir
b,
Syed
Khalid
a,
Song
Ma
a,
Xiaoyu
Li
a,
Yunjun
Luo
*a and
Mu-Hua
Huang
*a
aSchool of Materials Science and Engineering, Beijing Institute of Technology, Beijing, 100081, China. E-mail: yjluo@bit.edu.cn; mhhuang@bit.edu.cn
bDepartment of Chemistry, Government Samanabad College for Women, Faisalabad, Pakistan
First published on 8th January 2016
Abstract
To improve the thermo-mechanical properties of glycidyl azide polymer (GAP) and hydroxyl terminated polybutadiene (HTPB) based propellants, a facile sequential polymerization approach has been conducted to prepare an energetic hybrid polymer network (EHPN) through stepwise curation. The detailed curing conditions for the EHPN formation were determined using an in situ FTIR kinetic study. The effect of curing ratio (NCO/OH) on the mechanical properties of the polyurethane networks of GAP and HTPB was investigated, wherein hexamethylene diisocyanate biuret trimer (Desmodur N100) and isophorone diisocyanate (IPDI) were used as mixed curative agents. A series of EHPNs were prepared by varying the relative weight ratios of GAP and HTPB with a single poly-isocyanate mixed curing system (IPDI/N100). A remarkable mechanical strength of up to 5.83 MPa and an elongation at break of 359% were achieved with a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 weight ratio of GAP to HTPB, which is the maximum mechanical strength reported thus far for a binder system of GAP and HTPB, which has a thermally more stable cross-linked network. The thermal properties of the as-synthesized PU networks of GAP, HTPB and GAP–HTPB EHPNs with different weight ratios were characterized using the DMA and DSC techniques. Thermal degradation behavior and morphological studies were also investigated with TGA-DTG and scanning electron microscopy (SEM), respectively. The facile sequential polyurethane curation polymerization technique can be potentially used for advanced solid composite propellants.
:50 weight ratio of GAP to HTPB, which is the maximum mechanical strength reported thus far for a binder system of GAP and HTPB, which has a thermally more stable cross-linked network. The thermal properties of the as-synthesized PU networks of GAP, HTPB and GAP–HTPB EHPNs with different weight ratios were characterized using the DMA and DSC techniques. Thermal degradation behavior and morphological studies were also investigated with TGA-DTG and scanning electron microscopy (SEM), respectively. The facile sequential polyurethane curation polymerization technique can be potentially used for advanced solid composite propellants.
Introduction
State-of-the-art solid rocket propellants usually contain low-vulnerability binders, which are composed of oxidizers and pre-polymers/plasticizers with energetic groups such as –N3 (azido), nitro (C-nitro, O-nitro (nitrate ester) and N-nitro (nitramine)) and difluroamine groups. As a result, the internal energy of the product is enhanced as well as the overall oxygen balance.1 The polymeric binders in propellants are normally cross-linked polyurethane elastomeric networks, which function as a matrix to bind solid (such as oxidizers and metal fuels) plasticizers and other minor additives. Among the energetic polymers, azido polymers have drawn immense attention in the development of solid propellants. Glycidyl azide polymer (GAP) is an exclusive polymer of high density with the positive heat of formation of +117.2 kJ mol−1.2 It outperforms all other azido polymers that have been developed during the last decade, due to its positive effect on the specific impulse and burning rate of solid composite propellants through the exothermic C–N3 group scission reaction.3–6GAP is considered to be a distinctive contender binder for eco-friendly, chlorine-free and smokeless solid propellants.3,4 However, GAP-based propellants do not reveal good mechanical strength, especially at low temperatures, due to the high chain stiffness from its polyether backbone structure. Normally, the polymeric binder characteristics can significantly influence the structural integrity of the propellant. Many attempts have been made to overcome this problem via the co-polymerization of GAP with poly ethylene glycol (PEG), poly caprolactone (PCL), tetrahydrofuran (THF), ethylene oxide (EO) and HTPB, and their mechanical and thermal properties have been reported.7–11 For example, Min et al. found that the block co-polyurethane binder matrices of GAP/poly ethylene glycol and GAP/poly caprolactone had enhanced mechanical strength.7 The glass transition temperature of GAP (6 °C) is much greater than that of HTPB and this considerably limits its application in composite solid propellants. HTPB is widely used as a polymeric binder in composite propellants due to its excellent physico-chemical properties.11–14 HTPB presents a low glass transition temperature, hydrolytic stability, high flexibility and resistance to solvents, which make it ideal for composite solid propellants.15,16 Several research groups have combined HTPB with other pre-polymers to achieve better mechanical, thermal and chemical resistance properties. Interpenetrating polymer networks (IPNs) based on HTPB PU/poly(methyl methacrylate), HTPB-PU/polystyrene and HTPB-PU/poly(ethylene oxide) have been investigated.17–21
Many researchers have explored different approaches to improve the mechanical strength of GAP and HTPB, although they are significantly restrained by the poor compatibility between GAP and HTPB due to the polar nature of GAP and non polar nature of HTPB.22 Mathew et al. reported the synthesis of GAP–HTPB cross-linked networks and achieved the mechanical strength of around 4.2 MPa and elongation <200% with 30% GAP–MDCI (methylene bis cyclohexyl isocyanate) with two glass transition temperatures at −74.03 °C and −35.84 °C.23 Ding et al. used a triazole curing system based on propargyl-terminated polybutadiene (PTPB) and GAP and reported a maximum mechanical strength of up to 2.5 MPa and elongation around 50% with (N3:C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) molar ratios of 1:2.2 Bing et al. achieved 3.83 MPa tensile strength and 593% elongation using GAP–HTPB blend binders with 50:50 weight ratios but no thermal properties of the as-synthesized GAP–HTPB binder system were reported.24 Mohan et al. synthesized the copolymer of GAP and HTPB, which showed two glass transition temperatures, due to the incompatibility of their polymeric chains. Moreover, only thermal properties of the copolymer were investigated.8
C) molar ratios of 1:2.2 Bing et al. achieved 3.83 MPa tensile strength and 593% elongation using GAP–HTPB blend binders with 50:50 weight ratios but no thermal properties of the as-synthesized GAP–HTPB binder system were reported.24 Mohan et al. synthesized the copolymer of GAP and HTPB, which showed two glass transition temperatures, due to the incompatibility of their polymeric chains. Moreover, only thermal properties of the copolymer were investigated.8
In recent years, HPNs, which combine the properties of components by forming networks and hybrid polymerization, have attracted extensive attention and is an innovative approach to elucidate the problem of polymer incompatibility.25–31 Normally, two types of routes are used to form HPNs, namely, sequential and simultaneous polymerization. Sequential polymerization is generally the process in which a second polymeric component network is polymerized following the completion of polymerization of the first component network, whereas simultaneous polymerization is the process in which both component networks are polymerized concurrently.28–32 HPNs have gained more and more research attention because of their outstanding properties and the synergetic effect introduced by the forced compatibility of individual components, and thus the enhanced mechanical strength and resistance to thermal degradation, which result from the catenation and entanglements of polymer chains.33–38
We recently reported the preparation of energetic IPNs of azido–alkyne click and polyurethane using acyl-GAP and HTPB with dual a curing system (DDPM-IPDI/N100) via the “simultaneous” polymerization technique to achieve optimum mechanical strength and a thermally stable binder system for composite propellants.39 In order to further simplify the procedure while retaining the good mechanical properties, we constructed facile “sequential” EHPN networks of GAP and HTPB by stepwise curing with a single poly-isocyanate mixed curing system (IPDI/N100), as shown in Fig. 1. Comprehensive information regarding the curing conditions of GAP and HTPB at different temperatures was provided by an in situ FTIR kinetic study. Single and inward shifting of the glass transition temperatures (Tg) was shown by dynamic mechanical analysis (DMA) and differential scanning calorimetry (DSC) and the results from the thermo-gravimetric analysis (TGA-DTG) show that the network formed is thermally more stable. IPN with a weight ratio of 50:50 (GAP:HTPB) gave synergetic mechanical strength, and microscopic studies demonstrates the interlocking and entanglement of the GAP–HTPB networks. To the best of our knowledge, our technique for sequential EHPN synthesis is more facile and gives the maximum mechanical strength reported thus far for a binder system of GAP and HTPB, which has a thermally more stable cross-linked network.
 | ||
| Fig. 1 Schematic of the polyurethane reaction and EIPN structure of PU based on GAP–HTPB. | ||
Experimental section
Materials
GAP with a molecular weight of 3700 g mol−1 and hydroxyl content of 29.15 mg KOH g−1, nitrogen content of 41.3%, water content of 0.217% and functionality of 1.92 was purchased from the Liming Research Institute of Chemical Industry, Henan, China. HTPB with a molecular weight of 3020 g mol−1, hydroxyl content of 0.73 mmol g−1, hydrogen peroxide (H2O2) content of 0.022%, water content of 0.015% and functionality of 2.205 was also purchased from the same supplier and both GAP and HTPB were used after vacuum drying for 3 hours at 80 °C. Isophorone diisocyanate (IPDI) with an average molecular weight of 222.2 g mol−1 containing 9.0009 mmol of NCO per gram IPDI, hexamethylene diisocyanate biuret trimer (Desmodur N100) with the molecular weight of 725 g mol−1 and 5.379 mmol of NCO per gram and dibutyl tindilurate (DBTDL) 0.5% solution in diisooctyl sebacate (DOS) were purchased from Beijing Chemical Plant.Preparation of single networks
All the reagents were dried overnight in a vacuum oven at 60 °C before use. The polyurethane HTPB network was synthesized by mixing a stoichiometric amount of HTPB, IPDI and N100 in a beaker followed by degassing in a vacuum oven at 30 °C. The equivalent ratio of NCO/OH and IPDI/N100 weight ratio was 1.0. The required amount of DBTDL (0.3%) was added as a curing catalyst. The mixture was poured into a Teflon coated mold and degassed in a vacuum oven. The molds were finally cured at 70 °C for 5 days. The polyurethane GAP network was also prepared using the same procedure, except that HTPB was replaced by GAP.Synthesis of energetic hybrid polymer network (EHPNs)
GAP–HTPB EHPNs were synthesized via the “sequential” polymerization method, wherein the pre-polymers, curing agent, cross-linking agent and catalyst were mixed together (same proportions for each network as used in the single network preparation). A series of GAP–HTPB EIPNs were synthesized by varying the relative weight proportions of HTPB (90%, 70%, 50%, 30%, and 10%) with respect to GAP. The required amount of GAP, HTPB along with IPDI, N100 and DBTDL were placed in a beaker followed by degassing in a vacuum oven at 30 °C. The equivalent ratio of NCO/OH and IPDI/N100 weight ratio was also the same as in the single networks. The entire mixture was poured into a Teflon coated mold and degassed in a vacuum oven at 30 °C. The mold was cured at 30 °C for 3 days followed by 70 °C for 5 days for stepwise curing. A schematic of the reaction of GAP and HTPB with the single poly-isocyanate mixed curative system (IPDI/N100) is shown in Scheme 1. | ||
| Scheme 1 Schematic of the reaction of GAP and HTPB with the mixed curing system IPDI and N100. | ||
Measurement of mechanical and thermal characteristics
IR spectra were obtained on a Nicolet FTIR-8700 Thermo spectrophotometer in the range of 4000–500 cm−1. The mechanical properties, including tensile strength (σb) and elongation at break (εb), of all the dumbbell-shape specimens were determined using a universal testing machine (Instron-6022, Shimadzu Co., Ltd.) at a constant rate of 100 mm min−1 and the results were averaged from five samples. Glass transition temperatures (Tg) were obtained using differential scanning calorimetry (DSC Mettler Toledo DSC1) with a heating rate of 10 °C min−1 over a temperature range from −100 to 50 °C under a nitrogen flow of 40 mL min−1.Dynamic mechanical tests were performed on a DMA 242C (Netzsch, Hanau, Germany) with a dual cantilever device at a frequency of 1 Hz. The temperature range was from −100 to 50 °C under a nitrogen atmosphere with a heating rate of 3 °C min−1. The dimensions of the test specimens were 30 mm × 10 mm. Thermogravimetric analysis was performed on a TGA analyzer (TGA/DSC1SF/417-2, Mettler Toledo) at a scanning rate of 10 °C min−1 under a nitrogen atmosphere of (40 mL min−1) from room temperature (25 °C) to 600 °C. Scanning electron microscopy (SEM) S-4800 (Hitachi) was used to image the surfaces of the films. All the film samples were fractured with liquid nitrogen and the samples were coated with a thin layer of gold before imaging.
Results and discussion
In situ FTIR kinetic studies of GAP/IPDI-N100 and HTPB/IPDI-N100
In our previous study, it was confirmed that the mechanical strength and thermal properties of GAP could be improved by the incorporation of HTPB via in situ polymerization using a dual curing system to achieve the energetic acyl-GAP/HTPB EIPNs.39 In this study, we investigate the sequential polymerization of GAP and HTPB by controlling the curing conditions. Our group reported in situ FTIR kinetic studies of GAP/IPDI-N100 and HTPB/IPDI-N100.40,41 This provided us with the kinetic information for the curing reaction, which enabled us to adjust the curing conditions for the sequential polymerization of EHPN formation.Fig. 2 depicts the reaction kinetics of polyurethane formation in terms of distinctive peak conversion with respect to time. The PU kinetics was followed by observing the change in intensity of the absorption band of the NCO stretching at 2258 cm−1 and CO stretching at 1730 cm−1. The difference in the intensity of the peaks was obtained with the Omnic software. The intensity of the peak for the NCO stretching band decreased and finally disappeared upon completion of the reaction. The new absorption peaks at 1731 cm−1 and 1508 cm−1 were assigned to CO and NH stretching bands, respectively, which reveal the formation of PU.
 | ||
| Fig. 2 NCO conversion degree–time curves for GAP and HTPB with IPDI/N100 at 70 °C. | ||
In situ FTIR kinetic study shows that around 80% of the HTPB PU network formation is completed within 10 hours, whereas the GAP PU network formation needs almost 80 hours. All the hydroxyl groups of HTPB are primary, whereas GAP contains approximately 10% primary and 90% secondary hydroxyl groups. The reactivity of the primary hydroxyl groups is higher than that of secondary hydroxyl groups. Moreover, the reactivity of NCO groups from N100 (aliphatic) and IPDI (cyclic) is also different. The primary NCO groups of IPDI are almost ten times more reactive than the secondary groups. There are three primary NCO groups in Desmodur N100 that are more reactive with the hydroxyl groups of HTPB and GAP. Fig. 3 demonstrates the in situ FTIR kinetic study of GAP, HTPB and GAP–HTPB (50:50) at 30 °C. In the case of HTPB, almost 95% polyurethane formation takes place in three days, whereas only 6% NCO conversion takes place for GAP. In the case of the GAP–HTPB (50:50) mixed binder system at 30 °C, almost 52% NCO conversion takes place and this may be due to the reactivity differences of the hydroxyl groups of GAP and HTPB. This is why in the mixed binder system (50:50), almost 52% polyurethane (PU) formation takes place at 30 °C and the remaining NCO conversation takes place at a higher temperature (70 °C). Based on the reactivity differences of the hydroxyl and NCO groups of the pre-polymers and curing agents, in situ FTIR kinetic study motivated us to use sequential polymerization for GAP–HTPB EHPN formation. Although it does not follow 100% sequential polymerization, on the basis of stepwise curation, we can say that sequential polymerization takes place in the glycidyl azide polymer and hydroxyl terminated polybutadiene. During the first three days of curing at 30 °C, most of the HTPB PU network formed, whereas a modest amount of GAP PU network formed. The second step curing at 70 °C for five days ensures the complete PU network formation of GAP and HTPB. This stepwise curing enables the entanglement and cross-linking of both PU networks, and the effect of this catenation is studied in detail in the mechanical and thermal part of this manuscript.
 | ||
| Fig. 3 NCO conversion degree–time curves for GAP, HTPB and GAP–HTPB with IPDI/N100 at 30 °C. | ||
Mechanical properties
In order to study the influence of polymerization and cross-linking on GAP/IPDI-N100 and HTPB/IPDI-N100 curing and how they influence the mechanical properties of the binder system, we need to examine the curing system by performing a series of uniaxial tensile tests without solid loading and non-plasticized polymer samples. Fig. 4 and 5 show the stress strain data of GAP/IPDI-N100 and HTPB/IPDI-N100 at varying curing ratios (NCO/OH). These tests sequences function as a sort of reference for additional evaluations, as predicted. Mechanical strength was increased and elongation correspondingly decreased with an increase in the curing ratios. Fig. 4 shows that by increasing the curing ratio (NCO/OH) from 0.8 to 1.8 in GAP/IPDI-N100, tensile strength gradually increased from 0.35 to 0.93 MPa, whereas the breaking elongation decreased from 280% to 104%. Using an (NCO/OH) ratio of >1 in both the GAP and HTPB binder system, tensile strength gradually increased at the expense of elongation due to the higher cross-linked density, which restricts chain mobility.42 As a result of this, breaking elongation decreased and tensile strength increased by increasing the curing ratio (NCO/OH). The same trend was observed in the HTPB/IPDI-N100 curing system (Fig. 5) in which the tensile strength progressively increased from 1.33 to 2.11 MPa and elongation dropped from 590% to 267%. Fig. 6 depicts the dependence of tensile strength (σb) and elongation at break (εb) of GAP–HTPB EHPNs. Pure GAP and the HTPB urethane network show the tensile strength (σb) of 0.56 and 1.48 MPa, and breaking elongation (εb) of 194 and 463%, respectively. | ||
| Fig. 4 Effect of curing ratio (NCO/OH) on the tensile strength (σ) and breaking elongation (εb) of GAP. | ||
 | ||
| Fig. 5 Effect of curing ratio (NCO/OH) on the tensile strength (σ) and breaking elongation (εb) of HTPB. | ||
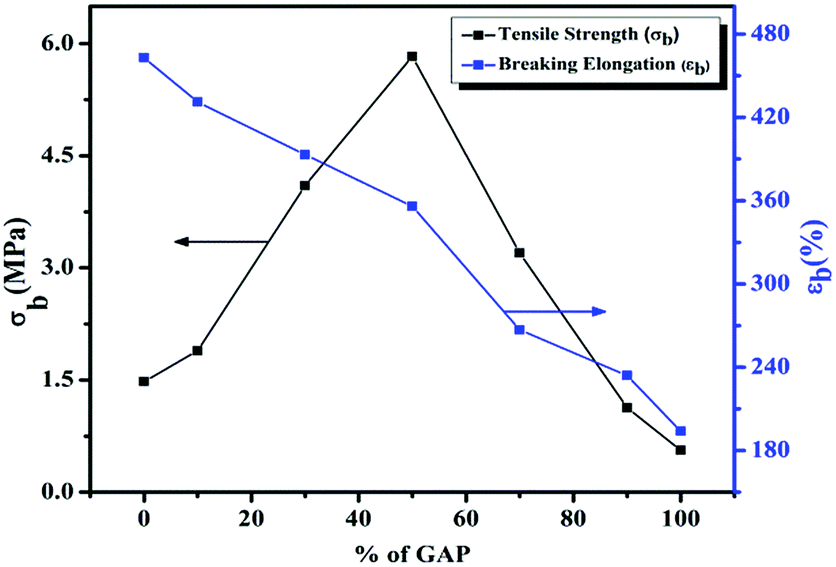 | ||
| Fig. 6 Effect of % of GAP on the tensile strength (σ) and breaking elongation (εb) of GAP–HTPB EHPNs. | ||
Herein, we also chose a curing ratio (NCO/OH) of 1.0. As we mentioned above, sequential polymerization was adopted for HPN formation. Fig. 6 shows that by increasing the weight ratio of GAP, tensile strength firstly increased from 1.48 MPa to 5.83 MPa and then steadily decreased to 0.56 MPa, but breaking elongation gradually decreased from 463% to 194%. An extensive increase in tensile strength and decrease in breaking elongation occurred during sequential polymerization. Fig. 1 and 2 (ESI†) clearly demonstrate the effect of curing ratio (NCO/OH) on the crosslinking density and swelling behavior of GAP and HTPB, respectively. Fig. 1 and 2 show almost the same trend of crosslinking density, wherein crosslinking density increased with the increase in NCO/OH ratio, whereas the swelling ratio decreased. The increase of crosslinking points can inhibit the swelling behaviour. With an NCO/OH ratio > 1 in both GAP and HTPB binder systems, tensile strength gradually increases at the expense of elongation due to formation of stiffer structures and more PU linkages occur between the hydroxyl groups of GAP and HTPB, which restrict chain mobility. As a result of this, the matrix takes more loads with less breaking elongation. In general, a higher crosslinking density results in higher tensile strength and lower elongation at break, and therefore with the increase in NCO/OH ratio, crosslinking density increased with an increase in tensile strength, whereas the breaking elongation decreased with the decrease in swelling ratio.
The tensile strength of 5.83 MPa with 359% elongation was achieved with 50:50% of GAP–HTPB EHPNs. A similar trend of crosslinking density was observed, whereas Fig. 3 (ESI†) also represents the maximum crosslinking with 50:50% GAP–HTPB EHPNs. This enhanced mechanical strength may be attributed to the hybrid network formation of GAP and HTPB. Due to catenation, chain flexibility is extremely restricted and elongation at break is considerably reduced. Beyond 50% GAP, the elasticity of GAP is inhibited by the azido groups and tensile strength and elongation at break gradually drop to 0.56 MPa and 194%, respectively, with 100% GAP network.
Thermal studies
δ and glass transition temperature (Tg) with weight ratio of GAP and HTPB.
 | ||
| Fig. 7 Variation of tanδ with temperature on (a) 0%, (b) 10%, (c) 30% (d) 50%, and (e) 70% GAP in GAP–HTPB EHPNs. | ||
Fig. 7 also shows that an inward shift of the glass transition temperature took place from −71 °C to −68 °C with a single glass transition temperature, whereas with 50:50 weight% of GAP–HTPB HPNs, two glass transitions were observed at −71 °C and −44 °C. The results from the DMA tests clearly show that HPNs with GAP up to 30% did not show phase separation with a single broad transition and maximum tanδ (1.05). Beyond 30%, the two glass transition temperatures might be due to phase separation. However, surprisingly synergetic mechanical strength was observed at 50:50 GAP–HTPB. Moreover, with 70% GAP, a single glass transition was observed and inward shifting of Tg and synergetic mechanical strength were observed. All these may be attributed to the interpenetration and entanglement of GAP and HTPB networks during sequential polymerization.
 | ||
| Fig. 8 TGA curves of GAP, HTPB and GAP–HTPB EHPNs. | ||
The residue after complete decomposition is around 1%. With an increase in the weight ratio of GAP, a thermally more stable network was formed and the decomposition temperatures increased, as clearly depicted in the DTG thermograms (Fig. 9). With 50% and 70% GAP weight ratios in GAP–HTPB EHPNs, the second stage decomposition increased to 501 °C and the peak decomposition temperatures also increased, as depicted in Fig. 9. The variation in decomposition may be ascribed to higher cross-linked networks and the resultant catenation. It can be seen from the DTG curves that during the first peak decomposition, the temperatures changed from 250 °C to 259 °C and from 458 °C to 468 °C during the second stage. Higher cross-linked networks need more energy for decomposition.2
 | ||
| Fig. 9 DTG curves of GAP, HTPB and GAP–HTPB EHPNs. | ||
:50% GAP–HTPB weight ratio shows two glass transition temperatures at −74 °C and −37 °C, which might be due to phase separation, but fortunately mechanical properties were enhanced. The same trend of inward shifting of Tg was observed for the 70% to 100% GAP cross-linked network. The characterization of impact, friction and electrostatic discharge sensitivities of the EHPNs and single networks of GAP and HTPB were performed using a standard procedure48 and the results clearly show that the single networks of GAP, HTPB and GAP–HTPB EHPNs were insensitive to impact, friction and ESD with >40 joules, >360 newton and having no ignition at 5.5 joules, respectively.
 | ||
| Fig. 10 DSC thermograms of HTPB, GAP and GAP–HTPB EHPNs. | ||
Morphological studies
Fig. 11 shows the morphological characteristics of the fractured surfaces of single networks of GAP, HTPB and GAP–HTPB EHPNs. The images of GAP and HTPB are shown in Fig. 11A and B, respectively. It can be seen that the fractured surface of a single network of GAP shows smooth and glassy microstructures, whereas HTPB shows rough, wrinkled and ravine microstructures. The SEM images of GAP–HTPB cross-linked networks (Fig. 11C and D) depict interlocking and entanglement due to the interpenetration of networks; wherein, polymer chains emerge to penetrate inward and outward over one another in the polymer matrix and thus show interlocking and a compact network with good compatibility.49–51 We also examined micro to nanometre thick long strands, which were crossed and entangled with each other. Synergetic mechanical strength, thermally more stable networks, inward shifting of Tg and increase of peak decomposition temperatures were significantly investigated during the sequential polymerization of GAP and HTPB with stepwise curing by controlling the temperature conditions. | ||
| Fig. 11 SEM images for the fractured surfaces of the films coded as (A) GAP PU (B) HTPB PU and (C and D) 50:50% GAP–HTPB PU EHPNs. | ||
Conclusions
The polyurethane curation of GAP and HTPB with isocyanate was comprehensively investigated by in situ FTIR, which resulted in the optimization of the polymerization conditions. Based on this, a facile sequential polymerization approach for the energetic hybrid polymer network (EHPN) formation of GAP and HTPB has been achieved by stepwise curation. Thermo-mechanical properties were investigated by the varying curing ratios (NCO/OH) for single networks of GAP and HTPB. By screening the GAP to HTPB weight ratio (1:9, 3:7, 5:5, 7:3 and 9:1) in the EHPN formation, superior mechanical characteristics have been achieved with a 50:50 GAP–HTPB weight ratio. DMA and DSC studies revealed that the inward shifting of the glass transition temperatures around 3–4 °C was observed with mostly single Tg and TGA-DTG studies demonstrated that the networks formed were thermally more stable. Moreover, peak decomposition temperatures increased by almost 10 °C. The morphological study illustrated that compact networks were formed due to the interlocking and catenation of both GAP and HTPB due to sequential polymerization.
References
- A. A. Provatas, Energetic Polymers and Plasticizers for Explosive Formulation—A Review of Recent Advances, DSTO-TR-0966, Defence Science & Technology Organization, Melbourne, Australia, 2000 Search PubMed.
- Y. Ding, C. Hu, X. Guo, Y. Che and J. Huang, J. Appl. Polym. Sci., 2014, 131, 40007 Search PubMed.
- K. Selim, S. Özkar and L. Yilmaz, J. Appl. Polym. Sci., 2000, 77, 538–546 CrossRef CAS.
- H. Kasıkçı, F. Pekel and S. Özkar, J. Appl. Polym. Sci., 2001, 80, 65–70 CrossRef.
- Y. Murali Mohan, M. Padmanabha Raju and K. Mohana Raju, J. Appl. Polym. Sci., 2004, 93, 2157–2163 CrossRef.
- S. K. Manu, T. L. Varghese, S. Mathew and K. N. Ninan, J. Appl. Polym. Sci., 2009, 114, 3360–3368 CrossRef CAS.
- B. Sun Min, Propellants, Explos., Pyrotech., 2008, 33, 131–138 CrossRef.
- Y. M. Mohan and K. M. Raju, Des. Monomers Polym., 2005, 8, 159 CrossRef CAS.
- A. Davenas, J. Propul. Power, 2003, 19, 1108–1128 CrossRef CAS.
- L. T. DeLuca, L. Galfetti, F. Maggi, G. Colombo, L. Merotto, M. Boiocchi, C. Paravan, A. Reina, P. Tadini and L. Fanton, Acta Astronaut., 2013, 92, 150–162 CrossRef CAS.
- M. K. Abkay and P. D. Davendra, Res. J. Chem. Environ., 2010, 14, 94–103 Search PubMed.
- D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, J. Hazard. Mater., 2008, 151, 289–305 CrossRef CAS PubMed.
- R. A. Assink, D. P. Lang and M. Celina, J. Appl. Polym. Sci., 2001, 81, 453–459 CrossRef CAS.
- K. Hailu, G. Guthausen, W. Becker, A. König, A. Bendfeld and E. Geissler, Polym. Test., 2010, 29, 513–519 CrossRef CAS.
- V. Sekkar, S. S. Bhagawan, N. Prabhakaran, M. Rama Rao and K. N. Ninan, Polymer, 2000, 41, 6773–6786 CrossRef CAS.
- H.-Q. Xie and J.-S. Guo, Eur. Polym. J., 2002, 38, 2271–2277 CrossRef CAS.
- C. Plesse, F. Vidal, C. Gauthier, J.-M. Pelletier, C. Chevrot and D. Teyssié, Polymer, 2007, 48, 696–703 CrossRef CAS.
- S. H. Wang, S. Zawadzki and L. Akcelrud, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 2861–2872 CrossRef CAS.
- M. Roha and F. Dong, J. Appl. Polym. Sci., 1992, 45, 1397–1409 CrossRef CAS.
- D. M. Jia, C. J. You, B. Wu and M. Z. Wang, Int. Polym. Process., 1988, 3, 205–210 CrossRef CAS.
- M. H. Han and S. C. Kim, Polym. Adv. Technol., 1997, 8, 741–746 CrossRef CAS.
- S. K. Manu, T. L. Varghese, S. Mathew and K. N. Ninan, J. Propul. Power, 2009, 25, 533–536 CrossRef CAS.
- S. Mathew, S. K. Manu and T. L. Varghese, Propellants, Explos., Pyrotech., 2008, 33, 146–152 CrossRef CAS.
- N. Bing, Q. G. Ming and R. Xiu-lun, Chin. J. Energ. Mater., 2010, 18(2), 167–173 Search PubMed.
- S. Ajithkumar, N. K. Patel and S. S. Kansara, Eur. Polym. J., 2000, 36, 2387–2393 CrossRef CAS.
- S. Chen, Q. Wang and T. Wang, Polym. Test., 2011, 30, 726–731 CrossRef CAS.
- P. Pissis, G. Georgoussis, V. A. Bershtein, E. Neagu and A. M. Fainleib, J. Non-Cryst. Solids, 2002, 305, 150–158 CrossRef CAS.
- I. Gitsov and C. Zhu, J. Am. Chem. Soc., 2003, 125, 11228–11234 CrossRef CAS PubMed.
- B. J. P. Jansen, S. Rastogi, H. E. H. Meijer and P. J. Lemstra, Macromolecules, 1999, 32, 6290–6297 CrossRef CAS.
- H. Nakanishi, M. Satoh, T. Norisuye and Q. Tran-Cong-Miyata, Macromolecules, 2004, 37, 8495–8498 CrossRef CAS.
- J. S. Turner and Y. L. Cheng, Macromolecules, 2000, 33, 3714–3718 CrossRef CAS.
- D. Gao and M. Jia, J. Appl. Polym. Sci., 2013, 128, 3619–3630 CrossRef CAS.
- Y.-Y. Hu, J. Zhang, Q.-C. Fang, D.-M. Jiang, C.-C. Lin, Y. Zeng and J.-S. Jiang, J. Appl. Polym. Sci., 2015, 132, 41537 Search PubMed.
- S. Chaudhary, S. Parthasarathy, D. Kumar, C. Rajagopal and P. K. Roy, J. Appl. Polym. Sci., 2014, 131, 40490 Search PubMed.
- J.-J. Wang, M.-B. Wu, T. Xiang, R. Wang, S.-D. Sun and C.-S. Zhao, J. Appl. Polym. Sci., 2015, 132, 41585 Search PubMed.
- F. Abbasi, H. Mirzadeh and A. A. Katbab, J. Appl. Polym. Sci., 2002, 86, 3480–3485 CrossRef CAS.
- O. Fichet, F. Vidal, J. Laskar and D. Teyssie, Polymer, 2005, 46, 37–47 CrossRef CAS.
- M. M. Jayasuriya and D. J. Hourston, J. Appl. Polym. Sci., 2012, 124, 3558–3564 CrossRef CAS.
- A. Tanver, M. Huang, Y. Luo, S. Khalid and T. Hussain, RSC Adv., 2015, 5, 64478–64485 RSC.
- S. Fei-Fei, A. Tanver and Y. Luo, Chin. J. Explos. Propellants, 2014, 37(4), 14–18 Search PubMed.
- A. Tanver, M. Huang, Y. Luo and Z. H. Hei, Adv. Mater. Res., 2015, 1, 337–341 Search PubMed.
- C. Hu, X. Guo, Y. Jing, J. Chen, C. Zhang and J. Huang, J. Appl. Polym. Sci., 2014, 131, 40007 Search PubMed.
- G. Herder, F. P. Weterings and W. P. C. de Klerk, J. Therm. Anal. Calorim., 2003, 72, 921–929 CrossRef CAS.
- E. L. M. Krabbendam-La Haye, W. P. C. de Klerk, M. Miszczak and J. Szymanowski, J. Therm. Anal. Calorim., 2003, 72, 931–942 CrossRef CAS.
- K. N. Ninan, K. Krishnan, R. Rajeev and G. Viswanathan, Propellants, Explos., Pyrotech., 1996, 21, 199–202 CrossRef CAS.
- J. K. Chen and T. B. Brill, Combust. Flame, 1991, 87, 217–232 CrossRef CAS.
- J. Simon and A. Bajpai, Eur. Polym. J., 2003, 39, 2077–2089 CrossRef CAS.
- C. Miró Sabaté, H. Delalu and E. Jeanneau, Chem.–Asian J., 2012, 7, 1085–1095 CrossRef PubMed.
- B. T. Low, T. S. Chung, H. Chen, Y. C. Jean and K. P. Pramoda, Macromolecules, 2009, 42, 7042 CrossRef CAS.
- B. J. P. Jansen, S. Rastogi, H. E. H. Meijer and P. J. Lemstra, Macromolecules, 1999, 32, 6290–6297 CrossRef CAS.
- S. Desai, I. M. Thakore, A. Brennan and S. Devi, J. Appl. Polym. Sci., 2002, 83, 1576–1585 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra23250c |
| This journal is © The Royal Society of Chemistry 2016 |