DOI:
10.1039/C5RA23192B
(Paper)
RSC Adv., 2016,
6, 43322-43329
Novel construction technique, structure and photocatalysis of Y2O2CN2 nanofibers and nanobelts†
Received
4th November 2015
, Accepted 25th April 2016
First published on 26th April 2016
Abstract
Y2O3 nanofibers and nanobelts were fabricated by calcination of the respective electrospun PVP/Y(NO3)3 composite nanofibers and nanobelts. For the first time, Y2O2CN2 nanofibers and nanobelts were successfully prepared via cyanamidation of the respective Y2O3 nanofibers and nanobelts employing NH3 gas and using graphite boat as container at 950 °C. X-ray power diffraction (XRD) analysis reveals that Y2O2CN2 nanofibers and nanobelts are pure trigonal phase with the space group of P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1. Scanning electron microscope (SEM) observation indicates that the diameter of Y2O2CN2 nanofibers is 167.59 ± 31.19 nm, and the thickness and width of Y2O2CN2 nanobelts are respectively 154 nm and 2.02 ± 0.84 μm under the 95% confidence level. Fourier transform infrared spectroscopy (FTIR) analysis manifests that the trigonal Y2O2CN2 nanofibers and nanobelts contain CN22− ions. Brunauer–Emmett–Teller surface area (BET) measurement shows the surface areas of the Y2O2CN2 nanofibers and nanobelts are 19.13 m2 g−1 and 15.92 m2 g−1, respectively. Y2O2CN2 nanostructures with different morphology exhibit high-efficiency photocatalytic capacity in photodegradation of rhodamine B (RhB) under the ultraviolet light irradiation, and the nanofibers have higher photocatalytic ability than nanobelts under the same experimental conditions. Furthermore, the nanofibers and nanobelts retain excellent photocatalytic stability after reused for four times. The photocatalytic mechanism and formation process of Y2O2CN2 nanofibers and nanobelts are also provided.
m1. Scanning electron microscope (SEM) observation indicates that the diameter of Y2O2CN2 nanofibers is 167.59 ± 31.19 nm, and the thickness and width of Y2O2CN2 nanobelts are respectively 154 nm and 2.02 ± 0.84 μm under the 95% confidence level. Fourier transform infrared spectroscopy (FTIR) analysis manifests that the trigonal Y2O2CN2 nanofibers and nanobelts contain CN22− ions. Brunauer–Emmett–Teller surface area (BET) measurement shows the surface areas of the Y2O2CN2 nanofibers and nanobelts are 19.13 m2 g−1 and 15.92 m2 g−1, respectively. Y2O2CN2 nanostructures with different morphology exhibit high-efficiency photocatalytic capacity in photodegradation of rhodamine B (RhB) under the ultraviolet light irradiation, and the nanofibers have higher photocatalytic ability than nanobelts under the same experimental conditions. Furthermore, the nanofibers and nanobelts retain excellent photocatalytic stability after reused for four times. The photocatalytic mechanism and formation process of Y2O2CN2 nanofibers and nanobelts are also provided.
1 Introduction
In recent years, rare-earth (RE) compounds have attracted much attention due to their applications in permanent magnets, superconductors, catalysts and optical devices, etc.1–3 There are many rare-earth compounds, such as RE2O2X (X = halogen, S2−, Se2−, Te2−, CO32−,4 CN22−,5,6 SO42−,7 etc.), in which the crystal structure of the rare-earth compounds consists of RE2O22+ layers and their interleaving anion. Different kinds of anions can make structure, physical and chemical properties unique and multiple. A wide variety of function is anticipated by changing interlayer anions between RE2O22+ layers.6 Research interest in C–N containing compounds of the lanthanides has been growing recently with the discovery of new (N–C–N)2− compounds such as Eu(CN2),8 LnCl(CN2)N (Ln = La, Ce),9 La2O(CN2)2 (ref. 10) and La3Cl(CN2)O3.11
Rare earth dioxymonocyanamides (Ln2O2CN2) have become a very important family of the rare earth C–N containing compounds. The structure and composition of La2O2CN2 powders were first reported by Hashimoto, et al.5 The linear anion (N–C–N)2− lies in parallel to the La2O22+ layers in structure because of the large ionic size of La3+. The La3+ ions are coordinated with 4 oxygen and 4 nitrogen atoms in tetragonal lattice.5 The (N–C–N)2− anions in RE2O2CN2 are perpendicular with the smaller RE3+, where RE = Y, Ce, Pr, Nd, Sm, Eu, Gd, Dy, Ho, Er, Tm, Yb. The RE3+ cations are coordinated with 4 oxygen and 3 nitrogen atoms in trigonal lattice.12–15
All the homologous Ln2O2(CN2) Ln = Y, Ce–Gd, Dy–Yb13–15 compounds were synthesized following the same procedure, solid-state metathesis (SSM) method or sol–gel method. For the first time, Xiaomin Guo with her co-workers successfully prepared tetragonal-phase La2O2CN2 nanofibers and nanobelts, which were synthesized through cyanamidation technique of La2O3 respective nanostructures at high temperature employing NH3 gas.16–19
Nanofibers and nanobelts are new kinds of one-dimensional nanostructures with special morphologies. They have attracted increasing interest of scientists owing to their anisotropy, large length-to-diameter ratio and width-to-thickness ratio, unique optical, electrical and magnetic performances.20–26 Therefore, study on the preparation and properties of nanofibers and nanobelts is still a popular subject in the field of materials science.
Electrospinning is a simple, convenient, and versatile technique to prepare long fibers with diameters ranging from tens of nanometers up to micrometers, including yttrium oxysulfide nanofibers and nanobelts,26,27 rare-earth yttrium oxyfluoride hollow nanofibers and composite oxide nanofibers and nanobelts.20,28–32 However, to the best of our knowledge, there have been no reports on the preparation of yttrium dioxymonocyanamides nanofibers and nanobelts by electrospinning combined with cyanamidation technique.
For Y, the ionic size (Y3+ = 0.1015 nm) might be too small to stabilize the oxycyanamide in trigonal structure,6 so, it is hard to prepare Y2O2CN2, and there are only a few reports about the preparation and structure of Y2O2CN2. Up to now, the fabrication of Y2O2CN2 nanofibers and nanobelts is not reported in the literature except for Y2O2CN2 powders.13,14 Herein, Y2O2CN2 nanofibers and nanobelts were fabricated by cyanamidation of the relevant Y2O3 nanostructures which were prepared by calcination of the electrospun nanostructures of PVP/Y(NO3)3 composites in an ammonia atmosphere using graphite boat at high temperature. The morphology, structure and photocatalytic properties of the resulting samples were investigated in detail, and the formation mechanisms of Y2O2CN2 nanostructures were also presented.
2 Experimental sections
2.1. Chemicals
Polyvinyl pyrrolidone (Mw = 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, AR) were purchased from Tianjin Bodi Chemical Co., Ltd. N,N-Dimethylformamide (DMF, AR) was bought from Tiantai Chemical Co., Ltd. Y2O3 (99.99%) was supplied by China Pharmaceutical Group Shanghai Chemical Reagent Company. Nitric acid (HNO3, AR) was bought from Beijing Chemical Co., Ltd. NH3 gas was supplied by Changchun Juyang Gas Co., Ltd. All chemicals were directly used as received without further purification.
000, AR) were purchased from Tianjin Bodi Chemical Co., Ltd. N,N-Dimethylformamide (DMF, AR) was bought from Tiantai Chemical Co., Ltd. Y2O3 (99.99%) was supplied by China Pharmaceutical Group Shanghai Chemical Reagent Company. Nitric acid (HNO3, AR) was bought from Beijing Chemical Co., Ltd. NH3 gas was supplied by Changchun Juyang Gas Co., Ltd. All chemicals were directly used as received without further purification.
2.2. Fabrication of Y2O2CN2 nanofibers
Y2O3 nanofibers were prepared by calcining the electrospun PVP/Y(NO3)3 composite nanofibers. 1.0000 g of Y2O3 powers were dissolved in dilute (1:1, volume ratio) HNO3 and evaporated to dryness by heating, then dissolved in 19.2347 g of DMF, and then 2.6782 g of PVP was added into the above solution under stirring for 8 h to form homogeneous transparent spinning solution. In the solution, the mass ratios of yttrium nitrate, DMF and PVP were 10:79:11. Subsequently, PVP/Y(NO3)3 composite nanofibers were prepared by electrospinning technique. The spinning solution was electrospun at a positive high voltage of 13 kV, distance between the capillary tip and the collector was 18 cm, and relative humidity was 10–40%. The collected electrospun composite nanofibers were then calcined at 700 °C in air for 8 h with the heating rate of 1 °C min−1 to obtain Y2O3 nanofibers.
The Y2O3 nanofibers were loaded into a graphite boat and then heated to 950 °C at a heating rate of 1 °C min−1 and remained for 12 h at 950 °C under a flow of gaseous ammonia. Then, the calcination temperature was decreased to 100 °C with a cooling rate of 1 °C min−1, followed by natural cooling down to room temperature, and Y2O2CN2 nanofibers were successfully obtained.
2.3. Synthesis of Y2O2CN2 nanobelts
Y2O3 nanobelts were prepared by calcining the electrospun PVP/Y(NO3)3 composite nanobelts. 1 g of Y2O3 were dissolved in dilute (1:1, volume ratio) HNO3 and evaporated to dryness by heating, then dissolved in 17.0434 g DMF, and then 4.8695 g PVP was added into the above solution under stirring for 12 h to form homogeneous transparent spinning solution. In the spinning solution, the mass ratios of yttrium nitrate, DMF and PVP were 10:70:20. Subsequently, PVP/Y(NO3)3 composite nanobelts were prepared by electrospinning technique. The spinning solution was electrospun at a positive high voltage of 8 kV, the distance between the capillary tip and the collector was 15 cm, and relative humidity was 40–60%. The collected electrospun composite nanobelts were then calcined at 700 °C in air for 8 h at a heating rate of 1 °C min−1 to acquire Y2O3 nanobelts.
Y2O2CN2 nanobelts were fabricated through cyanamidation of the obtained Y2O3 nanobelts using the same process, as described in Section 2.2.
2.4. Characterization methods
X-ray diffraction (XRD) analysis was performed using a Rigaku D/max-RA X-ray diffractometer with Cu kα radiation of 0.15406 nm. The size and morphology of the products were investigated by an XL-30 field emission scanning electron microscope (SEM) made by FEI Company. The purity of the products was examined by OXFORD ISIS-300 energy dispersive X-ray spectrometer (EDX). The specific surface areas of the nanostructures were measured by a V-Sorb 2800P specific surface area and pore size analyzer made by Gold APP Instrument Corporation. The samples were pre-evacuated for 120 min at 200 °C. Nitrogen gas is used as adsorption gas and adsorption process is carried out at 77 K applying nitrogen liquid as cooling agent. UV-Vis absorption spectra of the samples were taken with a UV-1240 spectrophotometer by Japanese Shimadzu Company. The Fourier transform infrared spectra of the samples were performed by a FTIR-8400S Fourier transform infrared spectrophotometer made by Shimadzu Corporation.
2.5. Evaluation of photocatalytic performance
In a typical photocatalytic reaction, 0.05 g of the as-prepared Y2O2CN2 nanostructures were dispersed into a 100 mL aqueous solution of RhB with the concentration of 0.1 g L−1. Prior to illumination, the mixture was stirring for 1 h in the dark to make the nanostructures evenly disperse and reach adsorption–desorption balance in solution. Then the solution was exposed directly under the ultraviolet light (500 W ultraviolet lamp with main emission wavelength of 365 nm, FHSDI F6T5-365) with stirring to trigger decomposition of the RhB molecules. In a 20 minute interval, 4 mL suspension was sampled and centrifuged to remove the photocatalyst powders. The concentration of RhB aqueous solution was analyzed at maximum absorption of 553 nm, using a Shimadzu UV-1240 UV-Vis spectrophotometer. The degradation rate of RhB by Y2O2CN2 nanostructures was estimated on the basis of the following formula:32
where A0 is the absorbance of RhB in the dark and A is the absorbance of RhB at given time intervals after irradiation.
The stability of the Y2O2CN2 nanostructures catalyst was evaluated by reusing the Y2O2CN2 nanostructures catalyst for four runs for the decomposition of RhB under the same conditions. After each run, the Y2O2CN2 nanostructures catalyst were centrifugally separated, then washed for four times using distilled water, and then they were respectively reused. All the experiments were performed at room temperature.
3 Results and discussion
3.1. XRD analysis
Fig. 1 demonstrates the XRD patterns of the Y2O3 nanostructures. As seen from Fig. 1, the characteristic diffraction peaks of samples are observed in 2θ range of 10–90°, which can be readily indexed to the cubic crystal phase of Y2O3 (PDF no. 25-1011).
 |
| | Fig. 1 XRD patterns of Y2O3 nanofibers (a) and Y2O3 nanobelts (b) with PDF standard card of Y2O3. | |
As there is no PDF standard card of Y2O2CN2, the phase composition of Y2O2CN2 nanofibers and nanobelts is confirmed by comparing the XRD patterns of the Y2O2CN2 products reported in the ref. 13. Fig. 2 shows the XRD patterns of the as-prepared Y2O2CN2 nanofibers and nanobelts. All the diffraction peaks are highly consistent those of the pure trigonal-phase of Y2O2CN2 powders with space group of Pm1,13 and their structure comprises distinct covalent Y2O22+ complex cation and CN22− anion layers. In this structure, the Y3+ ions are coordinated with four oxygen and three nitrogen atoms in trigonal lattice. Obvious diffraction peaks are situated near 2θ = 10.08°, 21.9°, 27.96°, 30.04°, 35.98°, 37.12°, 43.74°, 49.24°, 50.54°, 60.46°, 80°. No diffraction peaks of any other phases or impurities are detected, indicating that pure-phase Y2O2CN2 nanostructures are successfully prepared.
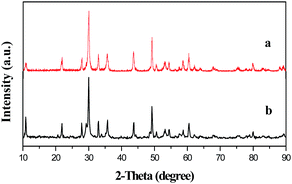 |
| | Fig. 2 XRD patterns of the Y2O2CN2 nanofibers (a) and Y2O2CN2 nanobelts (b). | |
3.2. Morphology observation
The morphologies of the products are characterized by scanning electron microscope (SEM). Fig. 3 manifests the representative SEM images of the composite nanofibers, composite nanobelts, Y2O2CN2 nanofibers and Y2O2CN2 nanobelts. From Fig. 3a, it can be noticed that the PVP/Y(NO3)3 composite nanofibers have smooth surface and uniform diameter. After annealing and cyanamidation at 950 °C, the diameter of the nanofibers greatly decreases due to loss of the PVP and associated organic components, as-formed Y2O2CN2 nanofibers have relatively rough surface and uniform diameter ranging from 100 nm to 300 nm, as revealed in Fig. 3b. The SEM image of PVP/Y(NO3)3 composite nanobelts with the thickness of 492 nm (shown in the inset of Fig. 3c) is manifested in Fig. 3c, the composite nanobelts are relatively smooth and uniform and the seeming variation in width is mainly due to the twist of a nanobelt: the width, thickness and the twist parts of a nanobelt are simultaneously shown in a SEM image. The actual width of a single nanobelt is almost unchanged. The width value of a nanobelt is obtained by measuring the widest section of the nanobelt. Clearly, uniform Y2O2CN2 nanobelts with the thickness of 154 nm (shown in the inset of Fig. 3d) are synthesized and have relatively rough surface, as indicates in Fig. 3d. Preliminarily, we can conclude that the cyanamidation plays an important role in keeping the morphology of the nanofibers and nanobelts.
 |
| | Fig. 3 SEM images of the composite nanofibers (a), Y2O2CN2 nanofibers (b), the composite nanobelts (c) and Y2O2CN2 nanobelts (d). | |
Under the 95% confidence level, the diameters of composite nanofibers and Y2O2CN2 nanofibers, the width of composite nanobelts and Y2O2CN2 nanobelts analyzed by Shapiro–Wilk method are normal distribution. Histograms of diameters and width of the nanostructures are indicated in Fig. 4. As seen from Fig. 4, the diameters of composite nanofibers, as-formed Y2O2CN2 nanofibers, the width of composite nanobelts and Y2O2CN2 nanobelts are 928.48 ± 96.06 nm, 167.59 ± 31.19 nm, 10.61 ± 2.92 μm, and 2.02 ± 0.84 μm, respectively.
 |
| | Fig. 4 Distribution histograms of diameter of the composite nanofibers (a), Y2O2CN2 nanofibers (b), the width of the composite nanobelts (c) and Y2O2CN2 nanobelts (d). | |
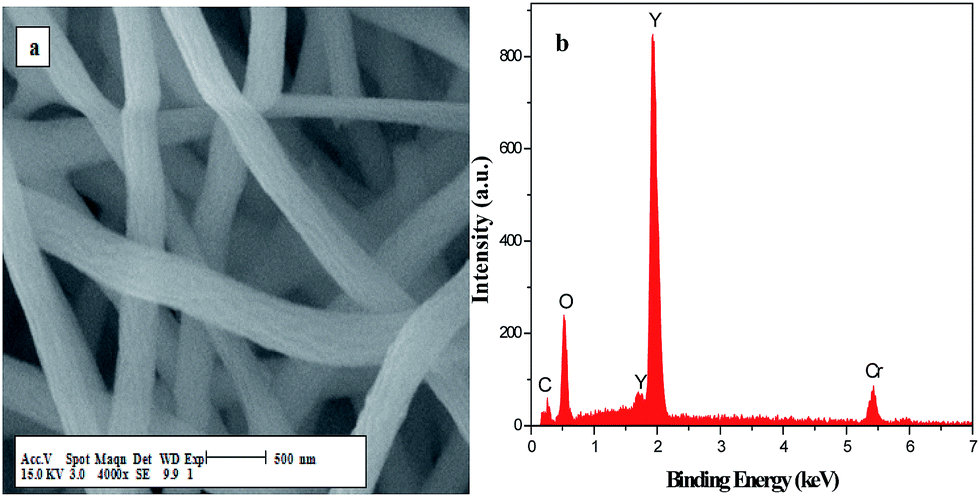
Fig. 5 demonstrates the EDX spectra of the PVP/Y(NO3)3 composites and Y2O2CN2 nanostructures. EDX spectra analysis show that C, N, O and Y are the main elements in PVP/Y(NO3)3 composites and Y2O2CN2 nanostructures. The content of C element in Y2O2CN2 nanostructures is much lower than that of the PVP/Y(NO3)3 composites due to the loss of the PVP and associate organic components. Au and Cr respectively come from the conductive films coated on the samples in preparation process for SEM analysis. No other elements are found in the samples, indicating that the Y2O2CN2 nanostructures are highly pure.
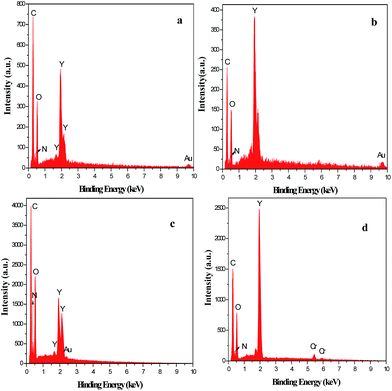 |
| | Fig. 5 EDX spectra of the composite nanofibers (a), Y2O2CN2 nanofibers (b), the composite nanobelts (c) and Y2O2CN2 nanobelts (d). | |
Fig. 6 respectively demonstrates the SEM image and elementary compositions of Y2O3 nanofibers. As reveals in Fig. 6b, the energy dispersive spectra reveal the presence of Y, O, C and N elements in Y2O3 nanofibers. The element of Cr in the spectrum comes from the Cr film coated on the surface of the sample for SEM observation. No other elements are found in the samples, indicating that the Y2O3 nanofibers are highly pure.
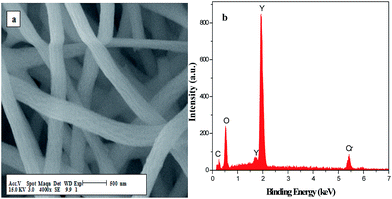 |
| | Fig. 6 SEM image (a) and EDX spectrum (b) of Y2O3 nanofibers. | |
3.3. FTIR spectra
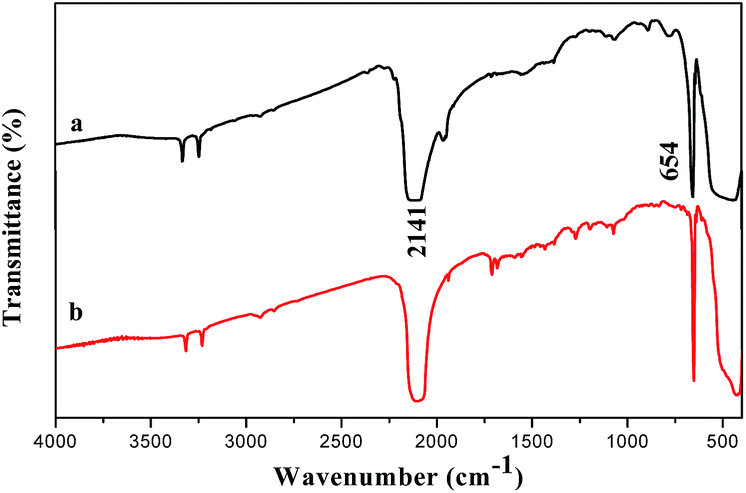
Fig. 7 shows the FTIR spectra of Y2O2CN2 nanostructures, they all show two main absorption peaks in the vicinity of 654 cm−1 and 2141 cm−1. These absorption peaks were assigned to the δ (formation vibration) and νas (antisymmetric stretch) modes of the CN22− ions in Y2O2CN2. The FTIR spectra of Y2O2CN2 indicates that the trigonal Y2O2CN2 nanofibers and nanobelts contain CN22− ions.12
 |
| | Fig. 7 FTIR spectra of Y2O2CN2 nanofibers (a) and nanobelts (b). | |
3.4. UV-Vis absorption spectra
Fig. 8 reveals the UV-Vis absorption spectra of the Y2O2CN2 nanofibers and nanobelts, the samples exhibit an intense absorption band in the range of 200–280 nm. It is well known that the optical absorption near the band edge for a direct transition crystalline semiconductor follows the formula:33,34 (αhν)1/2 = B(hν − Eg). Where α, h, ν, Eg and B are absorption coefficient, Planck constant, light frequency, band gap and a constant, respectively. The typical Eg of Y2O2CN2 nanofibers and nanobelts are respectively about 4.35 eV and 4.50 eV (the inset of Fig. 8; A is proportional to the absorption coefficient (α), α is substituted by A), which implies that the Y2O2CN2 nanofibers and nanobelts could be used as ultraviolet light active photocatalyst.
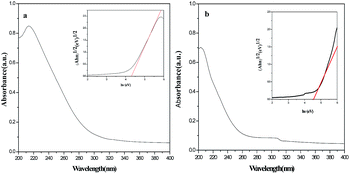 |
| | Fig. 8 UV-Vis absorption spectra of Y2O2CN2 nanofibers (a) and nanobelts (b). | |
3.5. Specific surface area analysis
The specific surface area of the Y2O2CN2 nanofibers and nanobelts were determined by BET method. The measured BET surface area adsorption–desorption isotherms of Y2O2CN2 samples are indicated in Fig. 9. The P/P0 ranges of Y2O2CN2 nanofibers and nanobelts were respectively 0.0120–0.3251 and 0.0097–0.3144. We conclude that the monolayer adsorption saturation capacity of nanofibers and nanobelts, which are obtained respectively from the inset of Fig. 9, are 2.37 mL and 2.81 mL. The BET model surface areas are calculated in the following formula:
| BET = [(Vm × 10−3)/22.4] × 6.02 × 1023 × 1.62 × 10−19 |
 |
| | Fig. 9 BET surface area adsorption isotherms of Y2O2CN2 nanofibers (a) and nanobelts (b). | |
The BET model surface areas of the Y2O2CN2 nanofibers and nanobelts are calculated to be 19.13 m2 g−1 and 15.92 m2 g−1 respectively. Remarkably, the specific surface area of the Y2O2CN2 nanofibers is bigger than that of the Y2O2CN2 nanobelts.
3.6. Photocatalysis
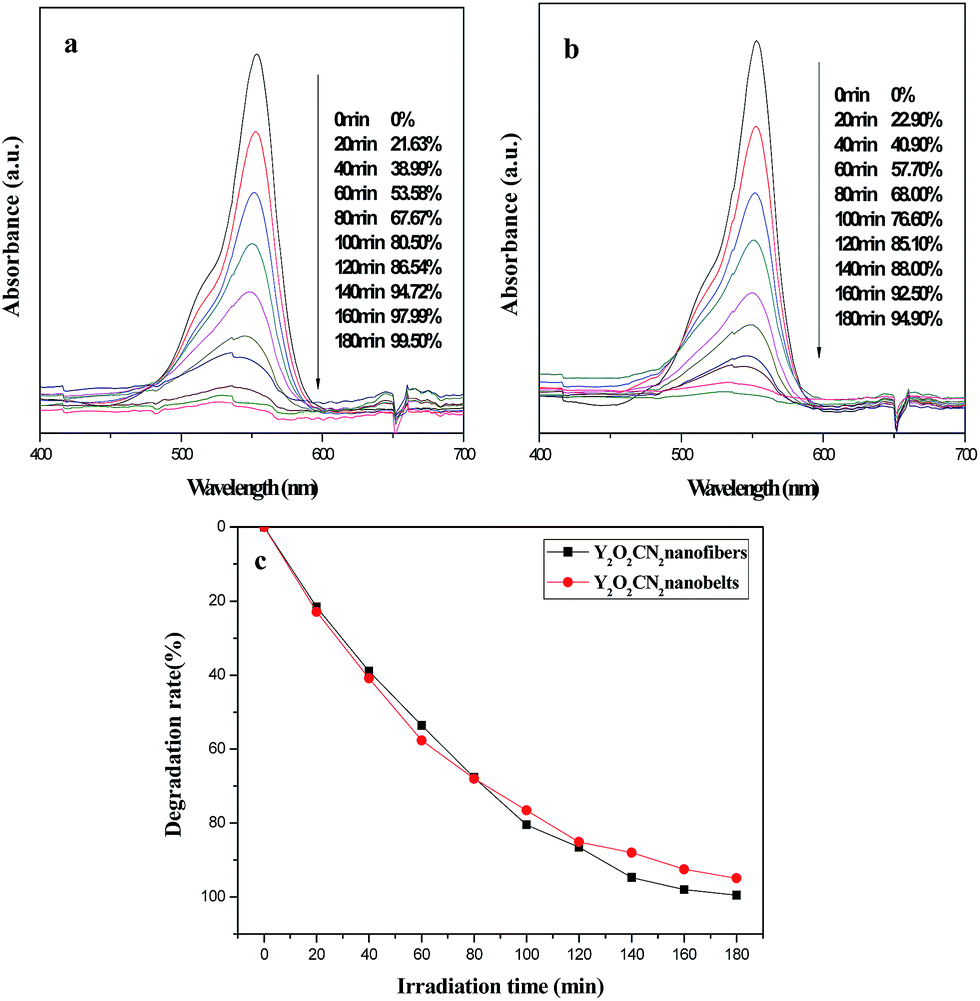
The photocatalytic activity of the Y2O2CN2 nanofibers and nanobelts were evaluated by the degradation of RhB aqueous solution under ultraviolet irradiation, as indicated in Fig. 10. Fig. 10a displays the absorption spectra of the RhB aqueous solution during the photocatalytic degradation process by the Y2O2CN2 nanofibers catalyst. The absorption spectra of RhB at λmax = 553 nm decrease gradually after photocatalytic reaction for 180 min, implying that the chromophoric groups of RhB molecules have been destroyed, and the degradation rates of RhB by Y2O2CN2 nanofibers and the Y2O2CN2 nanobelts were respectively 99.50% and 94.90%, as shown in Fig. 10a and b. Fig. 10c displays the degradation curves of RhB for the nanofibers and nanobelts at the first run. The rate constants for the degradation experiments are determined to be 0.0272 and 0.0165 for Y2O2CN2 nanofibers and nanobelts, respectively. The Y2O2CN2 nanofibers exhibit higher photocatalytic activities than the nanobelts from the beginning to the end, which is attributed to the fact that the BET surface area of nanofibers is bigger than that of the nanobelts. In order to confirm the stability of the photocatalytic performance of the Y2O2CN2 nanomaterials catalyst, the circulating runs in the photocatalytic degradation of RhB in the presence of Y2O2CN2 nanofibers and nanobelts catalysts under ultraviolet light are carried out, as shown in Fig. 11. After four recycles for the photocatalytic degradation of RhB, the catalysts do not exhibit significant loss of photocatalytic activity.
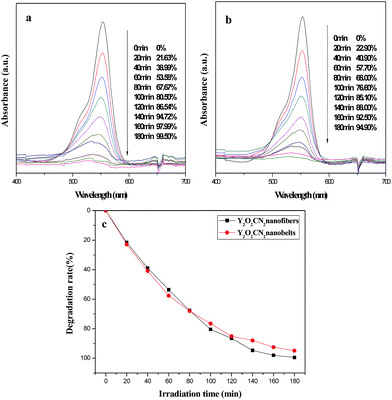 |
| | Fig. 10 Absorption spectra variation of RhB at different reaction time for Y2O2CN2 nanofibers (a) and Y2O2CN2 nanobelts (b), and the degradation curve of RhB over sample Y2O2CN2 nanomaterials (c). | |
 |
| | Fig. 11 Degradation curves of RhB over sample Y2O2CN2 nanofibers (a) and nanobelts (b). | |
Fig. 12 shows the first order reaction rate equation of the degradation experiments for Y2O2CN2 nanofibers and Y2O2CN2 nanobelts. The rate constants are determined to be 0.01685 min−1 and 0.01555 min−1 for Y2O2CN2 nanofibers and nanobelts.
 |
| | Fig. 12 The first order reaction rate curve and equation of the degradation experiments for Y2O2CN2 nanofibers (a) and Y2O2CN2 nanobelts (b). | |
3.7. Possible mechanism of the ultraviolet-induced photodegradation of RhB
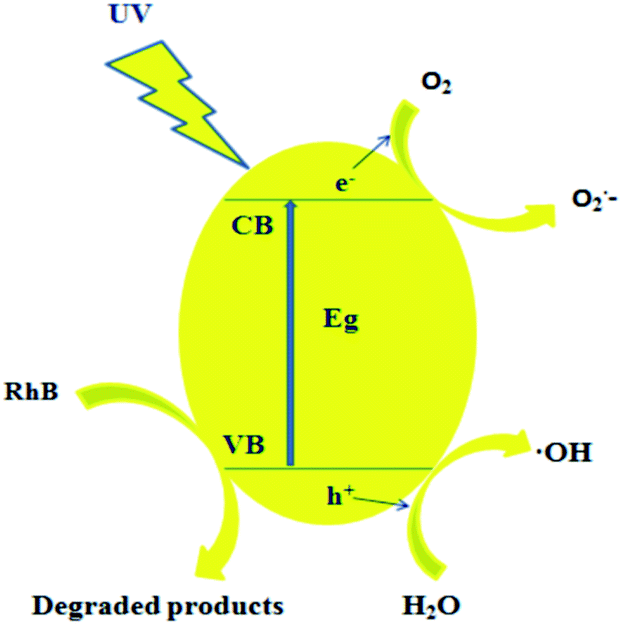
Based on the above results, a possible photocatalytic mechanism is indicated in Fig. 13. As shown in Fig. 13, Y2O2CN2 with narrow band gap energy (Eg of nanofibers and nanobelts are respectively about 4.35 eV and 4.50 eV) could be easily excited by ultraviolet light and the generation of photoelectrons and holes is induced. Then the photo-generated electrons (e−) probably react with dissolved oxygen molecules to yield super oxide radical anions, O2−, which on protonation generated the hydroperoxy, HO2˙, radicals, producing the hydroxyl radical OH˙, which is a strong oxidizing agent, to decompose the organic dye.35–40 A hypothetical mechanism is proposed for the photocatalytic degradation of RhB as follows:| | |
Y2O2CN2 + hν → Y2O2CN2(e− + h+)
| (1) |
| | |
Y2O2CN2(e−) + O2 → Y2O2CN2 + O2−
| (2) |
| | |
O2− + H2O → HO2˙ + OH−
| (3) |
| | |
HO2˙ + H2O → H2O2 + OH˙
| (4) |
| | |
Y2O2CN2(h+) + H2O + OH− → Y2O2CN2 + 2OH˙
| (6) |
| | |
OH˙ + RhB → degraded or mineralized products
| (7) |
 |
| | Fig. 13 Possible mechanism of the ultraviolet-induced photodegradation of RhB with Y2O2CN2 nanofibers and nanobelts. | |
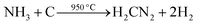
4 Formation mechanism for Y2O2CN2 nanostructures
According to the above analysis, we advance the formation mechanism of Y2O2CN2 nanostructures, as shown in Fig. 14. PVP and Y(NO3)3 are mixed with DMF to form spinning solution. PVP act as template during the formation of Y2O2CN2 nanostructures. Y3+ and NO3− are mixed or absorbed onto PVP to form PVP/Y(NO3)3 composite nanofibers and nanobelts via electrospinning. Some solvents are volatilized in the electrospinning process. PVP decompose soon and carbonize, nitrates are decomposed and oxidized to produce NO2, and eventually evaporate from the composite fibers and belts during calcinations process. With the increase in the calcining temperature, Y3+ could combine with O2, coming from air, to form Y2O3 crystallite, and many crystallites are combined into nanoparticles, and finally these nanoparticles mutually connect to generate Y2O3 nanofibers and nanobelts. Afterwards, the above products are cyanamidated in a graphite boat under the flowing NH3. In the cyanamidation process, the graphite from graphite boat reacts with the flowing ammonia gas and Y2O3 to produce Y2O2CN2, CO and H2 in the high temperature. During the process, graphite boat is not only a container, but also a reactant substance through reacting with NH3 and Y2O3 in the heating process. Cyanamidation technology we proposed here is actually a solid–gas reaction, which has been proved to be an important method, not only can retain the morphology of precursor, but also can fabricate pure phase Y2O2CN2 nanostructures. Reaction schemes for formation of Y2O2CN2 nanostructures proceed as follows:| |
 | (8) |
| |
 | (9) |
| |
 | (10) |
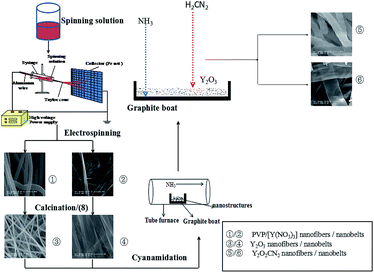 |
| | Fig. 14 Schematic diagram of formation mechanism of Y2O2CN2 nanofibers and nanobelts. | |
5 Conclusions
In summary, for the first time, pure trigonal phase Y2O2CN2 nanofibers and nanobelts with space group of Pm1 have been successfully prepared by electrospinning in conjunction with cyanamidation technology using NH3 gas and graphite at high temperature. The as-prepared Y2O2CN2 nanostructures have relatively rough surface, the diameter of the nanofibers is 167.59 ± 31.19 nm, the width of nanobelts is 2.02 ± 0.84 μm. Y2O2CN2 nanofibers and nanobelts possess excellent photocatalytic performance and their photocatalytic effects are stable. The cyanamidation technology we proposed here is of great importance, it can be applicable to the synthesis of other homologous rare earth oxycyanamide nanostructures with various morphologies such as Y2O2CN2:RE3+ and Ln2O2CN2:RE3+ (Ln = Pr, Nd, Gd, etc.).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51573023, 50972020, 51072026), Ph.D. Programs Foundation of the Ministry of Education of China (20102216110002, 20112216120003), the Science and Technology Development Planning Project of Jilin Province (Grant No. 20130101001JC, 20070402).
Notes and references
- M. Li, W. X. Yuan, J. F. Wang and C. Gu, Powder Diffr., 2007, 22, 59–63 CrossRef CAS.
- S. Q. Su, W. Chen, C. Qin, S. Y. Song, Z. Y. Guo, G. H. Li, X. Z. Song, M. Zhu, S. Wang, Z. M. Hao and H. J. Zhang, Cryst. Growth Des., 2012, 12, 1808–1815 CAS.
- J. B. Yu, L. Zhou, H. J. Zhang, Y. X. Zheng, H. R. Li, R. P. Deng, Z. P. Peng and Z. F. Li, Inorg. Chem., 2005, 44, 1611–1618 CrossRef CAS PubMed.
- G. G. Li, C. Peng, C. M. Zhang, Z. H. Xu, M. M. Shang, D. M. Yang, X. J. Kang, W. X. Wang, C. X. Li, Z. Y. Cheng and J. Lin, Inorg. Chem., 2010, 49, 10522–10535 CrossRef CAS PubMed.
- Y. Hashimoto, M. Takahashi, S. Kikkawa and F. Kanamaru, J. Solid State Chem., 1995, 114, 592–594 CrossRef CAS.
- T. Takeda, N. Hatta and S. Kikkawa, Chem. Lett., 2006, 35, 988–989 CrossRef CAS.
- M. Machida, K. Kawamura and K. Ito, J. Cheminf., 2004, 35, 662–663 Search PubMed.
- O. Reckeweg and F. J. DiSalvo, Z. Anorg. Allg. Chem., 2003, 629, 177–179 CrossRef CAS.
- R. Srinivasan, M. Ströbele and H.-J. Meyer, Inorg. Chem., 2003, 42, 3406–3411 CrossRef CAS PubMed.
- R. Srinivasan, S. Tragl and H.-J. Meyer, Z. Anorg. Allg. Chem., 2005, 631, 719–722 CrossRef CAS.
- M. Kubus, D. Enseling, T. Justel and H.-J. Meyer, Eur. J. Inorg. Chem., 2013, 2013, 3195–3199 CrossRef CAS.
- Y. Hashimoto, M. Takahashi, S. Kikkawa and F. Kanamaru, J. Solid State Chem., 1996, 125, 37–42 CrossRef CAS.
- J. Sindlinger, J. Glaser, H. Bettentrup, T. Jüstel and H.-J. Meyer, Z. Anorg. Allg. Chem., 2007, 633, 1686–1690 CrossRef CAS.
- M. Zeuner, S. Pagano and W. Schnick, Chemistry, 2008, 14, 1524–1531 CrossRef CAS PubMed.
- S. Pagano, M. Zeuner, U. Baisch and W. Schnick, Z. Anorg. Allg. Chem., 2010, 636, 2212–2221 CrossRef CAS.
- X. M. Guo, X. T. Dong, J. X. Wang, W. S. Yu and G. X. Liu, Chem. Eng. J., 2014, 50, 148–156 CrossRef.
- X. M. Guo, W. S. Yu, X. T. Dong, J. X. Wang, Q. L. Ma, G. X. Liu and M. Yang, J. Am. Ceram. Soc., 2015, 98, 1215–1222 CrossRef CAS.
- X. M. Guo, J. X. Wang, X. T. Dong, W. S. Yu and G. X. Liu, CrystEngComm, 2014, 16, 5409 RSC.
- X. M. Guo, W. S. Yu, X. T. Dong, J. X. Wang, Q. L. Ma, G. X. Liu and M. Yang, Eur. J. Inorg. Chem., 2015,(3), 389–396 CrossRef CAS.
- J. X. Wang, H. R. Che, X. T. Dong, L. Liu and G. X. Liu, Acta Opt. Sin., 2010, 30, 473–479 CrossRef CAS.
- Q. Z. Cui, X. T. Dong, J.
X. Wang and M. Li, J. Rare Earths, 2008, 26, 664–669 CrossRef.
- L. Y. Yang, J. X. Wang, X. T. Dong, G. X. Liu and W. S. Yu, J. Mater. Sci., 2013, 48, 644–650 CrossRef CAS.
- Q. L. Ma, J. X. Wang, X. T. Dong, W. S. Yu and G. X. Liu, Chem. Eng. J., 2013, 222, 16–22 CrossRef CAS.
- W. W. Ma, X. T. Dong, J. X. Wang, W. S. Yu and G. X. Liu, J. Mater. Sci., 2013, 48, 2557–2565 CrossRef CAS.
- Q. L. Ma, J. X. Wang, X. T. Dong, W. S. Yu, G. X. Liu and J. Xu, J. Mater. Chem., 2012, 22, 14438–14442 RSC.
- H. Y. Wang, Y. Yang, Y. Wang, Y. Y. Zhao, X. Li and C. Wang, J. Nanosci. Nanotechnol., 2009, 9, 1522–1525 CrossRef CAS PubMed.
- X. Lu, M. Yang, L. Y. Yang, Q. L. Ma, X. T. Dong and J. Tian, J. Mater. Sci.: Mater. Electron., 2015, 26, 4078–4084 CrossRef CAS.
- Y. Liu, J. X. Wang, X. T. Dong and G. X. Liu, Chem. J. Chin. Univ., 2010, 31, 1291–1296 CAS.
- X. T. Dong, L. Liu, J. X. Wang and G. X. Liu, Chem. J. Chin. Univ., 2010, 3, 20–25 Search PubMed.
- D. Li, W. S. Yu, X. T. Dong, J. X. Wan and G. X. Liu, J. Fluorine Chem., 2013, 145, 70–76 CrossRef CAS.
- D. Li, X. T. Dong, W. S. Yu, X. Wan and G. X. Liu, J. Mater. Sci.: Mater. Electron., 2013, 24, 3041–3048 CrossRef CAS.
- J. X. Wang, Y. Q. Guo, X. T. Dong, Z. G. Li and G. X. Liu, J. Inorg. Mater., 2010, 25, 379–385 CrossRef CAS.
- Y. C. Chou, C. L. Shao, X. H. Li, C. Y. Su, H. C. Xu, M. Y. Zhang, P. Zhang, X. Zhang and Y. C. Liu, Appl. Surf. Sci., 2013, 285, 509–516 CrossRef CAS.
- C. H. Wang, C. L. Shao, Y. C. Liu and X. H. Li, Inorg. Chem., 2009, 48, 1105–1113 CrossRef CAS PubMed.
- Z. Y. Zhang, C. L. Shao, X. H. Li, Y. Y. Sun, M. Y. Zhang, J. B. Mu, P. Zhang, Z. C. Guo and Y. C. Liu, Nanoscale, 2013, 5, 606–618 RSC.
- M. Y. Zhang, C. L. Shao, J. B. Mu, X. M. Huang, Z. Y. Zhang, Z. C. Guo, P. Zhang and Y. C. Liu, J. Mater. Chem., 2012, 22, 577 RSC.
- M. Y. Zhang, C. L. Shao, P. Zhang, C. Y. Su, X. Zhang, P. P. Liang, Y. Y. Sun and Y. C. Liu, J. Hazard. Mater., 2012, 225–226, 155–163 CrossRef CAS PubMed.
- M. Y. Zhang, C. L. Shao, Z. C. Guo, Z. Y. Zhang, J. B. Mu, T. P. Cao and Y. C. Liu, ACS Appl. Mater. Interfaces, 2011, 3, 369–377 CAS.
- P. Zhang, X. H. Li, C. L. Shao and Y. C. Liu, J. Mater. Chem. A, 2015, 3, 3281–3284 CAS.
- M. Y. Zhang, C. L. Shao, X. Zhang and Y. C. Liu, CrystEngComm, 2015, 17, 7276–7282 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra23192b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.