
Density functional theory approach to CO2 adsorption on a spinel mineral: determination of binding coordination
Hye Sook Moon†
a,
Soonchul Kwon†b,
Sung Hyun Kwona,
Min Cho*c,
Jeong Gil Seo*d and
Seung Geol Lee*a
aDepartment of Organic Material Science and Engineering, Pusan National University, Busandaehak-ro 63beon-gil, Geumjeong-gu, Busan 46241, Republic of Korea. E-mail: seunggeol.lee@pusan.ac.kr; Tel: +82-51-510-2412
bSchool of Urban, Architecture and Civil Engineering, Pusan National University, Busandaehak-ro 63beon-gil, Geumjeong-gu, Busan 46241, Republic of Korea
cDivision of Biotechnology, Advanced Institute of Environment and Bioscience, College of Environmental and Bioresource Sciences, Chonbuk National University, Iksan 54596, Republic of Korea. E-mail: cho317@jbnu.ac.kr; Tel: +82-63-850-0845
dDepartment of Energy Science and Technology, Myongji University, 116 Myongji-ro, Cheoin-gu, Yongin-si, Gyeonggi-do 17058, Republic of Korea. E-mail: jgseo@mju.ac.kr; Tel: +82-31-324-1338
First published on 11th March 2016
Abstract
The mechanism of the adsorption of CO2 onto various sites of MgAl2O4 (100), in particular with regards to binding coordination, was investigated using density functional theory (DFT) calculations. Of the available sites, CO2 binding was calculated to be strongly adsorbed to oxygen atoms on the octahedral Al3+ and tetrahedral Mg2+ sites, with adsorption energy values of −1.60 eV and −1.86 eV, respectively, which was attributed to the small band gap of the CO2–MgAl2O4 system. It is clearly found that strongly adsorbed CO2 molecules bound to MgAl2O4 using polydentate (e.g., bidentate and tridentate) bonds. We also simulated the adsorption of multiple CO2 molecules on MgAl2O4, and found three of eight CO2 molecules to be strongly adsorbed using tridentate bonds onto the MgAl2O4 surface, with an interaction energy of −0.61 eV. The other five CO2 molecules were also adsorbed, but weakly, i.e., using physical interactions with a modest binding energy of <0.10 eV and at a relatively long distance from the MgAl2O4 surface.
Introduction
Anthropogenic emissions from the combustion of fossil fuels account for a significant proportion of the global emissions of CO2, which is the main factor driving global warming.1 To stem such emissions from power plants, the generated CO2 must be separated from the flue gas and sequestered,2 and such CO2 capture and storage is a promising approach for slowing down the rate of increase of CO2 levels in the atmosphere.3 Of the various CO2 separation technologies, the application of solid adsorbents holds particular promise because this process requires relatively little energy to operate and has negligible corrosion problems with good recyclability and a wide operating temperature range, and has led to the development of economical and effective CO2 capture materials.3,4 Simple alkali-earth metal oxide minerals (e.g., MgO, CaO, and BaO) have been used as CO2 adsorbents due to their simplicity and low cost.5 In the carbonation process, these alkali-earth metal oxides react with CO2 exothermically, and the basicity of the metal oxides determines the reactivity with CO2.6 Furthermore, stable carbonates are produced by the carbonation process and can be utilized in environmentally friendly ways such as mine reclamation and soil amendments.7 However, the poor stability of carbonates at high generation temperatures has limited their practical applications.8 To improve their stability, other metal oxides such as Al2O3, SiO2, TiO2 and ZrO have been commonly added to alkali-earth metal adsorbents.9 We recently introduced the Mg-based spinel mineral magnesium aluminate (MgAl2O4) for CO2 adsorption on account of its high basicity,10 but the mechanism by which CO2 adsorbs onto MgAl2O4 has not yet been fully investigated using a theoretical approach. MgAl2O4 is a well-known refractory oxide used as a structural ceramic that shows high resistance to most acids and alkalis with a low electrical loss. These properties allow MgAl2O4 to be used in a wide range of applications in structural, chemical, optical and electrical industries.For research involving interaction energetics at the atomic level, quantum chemical density functional theory (DFT) calculations have been widely performed with high accuracy to determine intrinsic energetic properties such as adsorption energy with optimized geometry.11 Herein, we investigated the mechanism of CO2 adsorption on MgAl2O4 (100) by analyzing the adsorption energy of this process and the optimized geometry using DFT calculations. To obtain a fundamental understanding of such CO2 adsorption, we carried out the calculations for various orientations of CO2 bound at different sites and for the adsorption of multiple CO2 molecules on the MgAl2O4 (100) surface. These calculations can help us understand the binding coordination and accompanying characteristics of the most stable number of bound CO2 molecules due to charge redistribution between CO2 and MgAl2O4, because the interaction of CO2 molecules with the metal-oxide surface influences the adsorptive characteristics of the metal oxide.
Experimental
Computational details
Our calculations were performed using the DMol3 module of Materials Studio from Biovia with the generalized gradient approximation (GGA) and Perdew–Burke–Ernzerhof (PBE) functional.12 We used all-electron Kohn–Sham wave functions and DNP numerical basis with DFT-D3 correction13 for all DFT calculations. The GGA-PBE functional has been widely used to describe the interactions between molecules14 and minerals.15 For obtaining the optimized unit structures, we fully relaxed the unit structure of MgAl2O4, and four atomic layers were cleaved along the (100) crystallographic direction with the constraint of the fourth layer. Three-dimensional periodic slab models were employed to simulate infinite surfaces. On the supercell, a sorbent surface was exposed as a slab, providing absorption sites. Vacuum separation was set to 20 Å along the z direction to avoid interactions beyond the periodic boundary condition. The k-point sampling for the Brillouin zone was performed using a 4 × 4 × 1 Monkhorst–Pack k-point mesh.16 A self-consistent field (SCF) convergence set to 1 × 10−5 Ha. Adsorption energy, Eads, between the adsorbate and the adsorbent slab was determined by three individual energy calculations: (i) energy from energy minimized adsorbate, ECO2, (ii) energy of the optimized adsorbent slab without adsorbates, EMgAl2O4, and (iii) energy from geometry optimization of the adsorbent slab interacting with adsorbates, EMgAl2O4+CO2.
 | (1) |
Results and discussion
Properties of the spinel MgAl2O4 structure
MgAl2O4 spinel forms a cubic (isometric) close-packed (face-centered cubic, fcc) crystal phase with Al3+ and Mg2+ occupying octahedral and tetrahedral sites in the lattice. Fig. 1a shows the unit cell of the optimized structure, which yielded a cubic lattice length of 8.08 Å. This calculated lattice length is in good agreement with the 8.05 Å experimentally determined length.17 The Mulliken charge distribution analysis of MgAl2O4 indicated a (negative) charge of −1.33e on each O, and (positive) charges of 1.41e and 1.95e on each Mg and Al, respectively, which can provide sufficiently high basicity for promoting adsorption of CO2. Compared to that in primitive cubic structures such as MgO (atomic packing factor of 0.52), more tightly packed arrangement of the atoms in the fcc structure (atomic packing factor of 0.74) also leads to a greater attraction between the atoms since a crystal field stabilization energy of MgAl2O4 is generated by the σ-type interactions between the metal cations and the oxide anions in the solids.18 This description provides an explanation for the high adsorptive affinity of MgAl2O4 for introduced CO2 gas. | ||
| Fig. 1 Optimized crystal structure of MgAl2O4. (a) Structure of the unit cell of MgAl2O4. (b) The structure of a periodic slab of MgAl2O4; (c) top view of the slab structure. | ||
Using the optimized unit cells, three-dimensional periodic slab models of MgAl2O4 were developed to simulate “infinite” surfaces, as shown in Fig. 1b and c. Note that the experimental X-ray diffraction (XRD) patterns of MgAl2O4 (JCPDS no. 21-11052) indicate that, of the experimentally observed (400), (511), and (440) crystalline plains, the (100) plain has a readily available surface for CO2 adsorption since the (400) plain is observed clearly with a high-intensity first-order peak. Thus, four atomic layers were cleaved along the MgAl2O4 (100) crystallographic direction with the constraint of the fourth layers.
Adsorption of a single CO2 molecule on MgAl2O4
Eight oxygen sites are available on the MgAl2O4 (100) surface for the adsorption of a single CO2 molecule (Fig. 2a and b). Since spinel MgAl2O4 is symmetric structure, we focused on two differently oriented oxygen sites, with “site 1” corresponding to oxygen bound to an octahedral Al3+ site and “site 2” to a tetrahedral Mg2+ site. We first optimized the geometry of the CO2 adsorbed on MgAl2O4 (Fig. 2c and d). The geometry of the adsorbed CO2 molecule was found to be significantly different from that of the free CO2 molecule. Specifically, the OCO2–CCO2–OCO2 bond angle contracted from 180° to 134.1° when adsorbed onto site 1, and to 125.3° on site 2, while the CCO2–OCO2 bond length, whether for CO2 bound to site 1 or to site 2, increased from 1.18 Å to 1.26 Å, as shown in Fig. 2c and d, and Table 1. This result is consistent with the high adsorption energy at site 2 (−1.88 eV) compared to that at site 1 (−1.60 eV). Furthermore, the bond lengths between the adsorbed CO2 and OMgAl2O4 (i.e., the adsorption site on the MgAl2O4 surface) were calculated to be 1.99 Å and 2.16 Å at site 1 and 1.41 Å at site 2. Interestingly, at O site 1, CO2 was adsorbed using the bidentate bond type and at site 2 with the tridentate bond type. These results show that, as the CO2 molecule approached the oxygen atom of the MgAl2O4 surface, the linear molecular structure of free CO2 became bent into a V shape. The elongated CCO2–OCO2 bond length is also in agreement with a hybridization change from sp1 to sp2, which is consistent with a loss of a shared pair of electrons from each of the chemical bonds. In addition, the adsorption energy was calculated to be quite high (>−1.60 eV) for each site, which suggests the formation of a new chemical bond between CO2 and the MgAl2O4 surface and hence a strong chemisorption. The charge reorganization of the MgAl2O4 (100) surface and the adsorbed CO2 molecule was quantified as shown in Fig. 2. It is found that 0.64e and 0.67e charge was transferred from the MgAl2O4 (100) surface to single CO2 molecule for site 1 and site 2, respectively. | ||
| Fig. 2 Available oxygen sites on the MgAl2O4 (100) surface: (a) top view, and (b) tilted view. Optimized geometry of CO2 adsorption on (c) site 1 and (d) site 2. C.T. denotes the charge transfer (e) from the MgAl2O4 (100) surface to CO2 molecule. | ||
| Bond length (CCO2–OMgAl2O4) (Å) | Bond length (OCO2–CCO2) (Å) | Bond angle (OCO2–CCO2–OCO2) (°) | Adsorption energy (eV) | Bond type | |
|---|---|---|---|---|---|
| Site 1 | 1.99, 2.16 | 1.26 | 134.1 | −1.60 | Reverse bidentate |
| Site 2 | 1.41, 1.98 | 1.26 | 125.3 | −1.88 | Tridentate |
The strong chemical bond is attributed to the sharing of electrons between the adsorbed CO2 molecule and the MgAl2O4 surface. To determine the mechanism of this favorable adsorption, we analyzed energy trends on the basis of molecular orbitals. Fig. 2 also shows the energetics of the bands and the localization of the highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO) in the CO2–MgAl2O4 system. These features indicate the orbital energies and the size of the HOMO–LUMO gap in MgAl2O4 with CO2 adsorbed. The stable pristine MgAl2O4 showed a large band gap without CO2 adsorption. Adsorption of CO2 onto octahedral Al3+ (site 1) and tetrahedral Mg2+ (site 2) caused, according to the calculations, different changes in the molecular orbitals: there was a reduction of the band gap at the tetrahedral Mg2+ site to 0.12 eV, attributed to a change in the energy levels due to overlapping p orbitals and increasing electron delocalization compared to that of the octahedral Al3+ site, for which the band gap was calculated to be 0.36 eV. The low calculated band gap values suggest that relatively little energy is required to excite the molecule, and hence promote the interaction of CO2 with MgAl2O4.
Note that the orientation of the adsorbed CO2 molecule relative to the MgAl2O4 surface depends on which oxygen the CO2 is being adsorbed onto since the geometries of the exposed surfaces of the octahedral or tetrahedral sites for CO2 adsorption differ from one another.
In addition, we determined an energy profile of the corresponding optimized geometry as a function of reaction coordinate for the adsorption of CO2 to the tetrahedral Mg2+ site of MgAl2O4 (Fig. 3). To determine the energy profile of the adsorption of CO2, we optimized the structure with fixed carbon atom of CO2 at each reaction coordination. Note that the relative energy was calculated as the difference between the adsorption energy of a CO2 molecule located 4.0 Å from the surface and the adsorption energy at the evaluated distance from the surface. In the calculations, the CO2 molecule began to form appreciable attractive interactions with MgAl2O4, i.e., with a relative energy of −0.29 eV, at a distance of 2.60 Å from the MgAl2O4 surface. A re-hybridized CO2 formed at 1.42 Å from the MgAl2O4 surface with a relative energy of −1.78 eV.
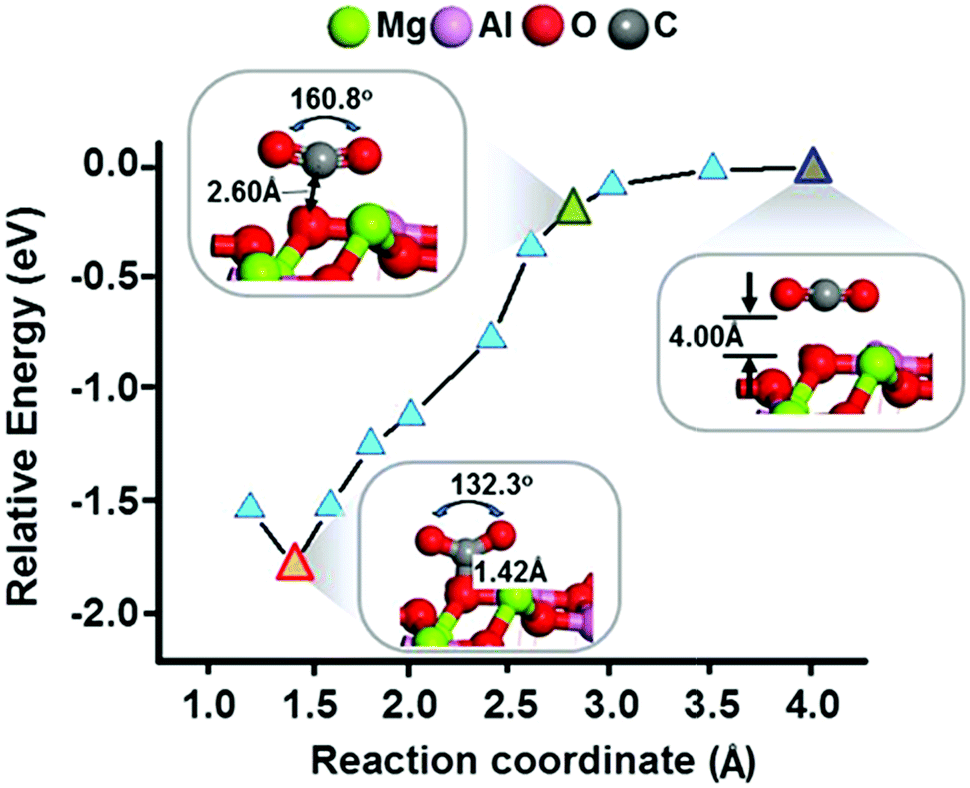 | ||
| Fig. 3 The energy profile of the adsorption of CO2 as it is moved from a vacuum to the MgAl2O4 (100) surface concurrent. The optimized geometries of this adsorption are shown at three locations on this profile. | ||
Adsorption of multiple CO2 molecules on MgAl2O4
Since many oxygen sites on the MgAl2O4 (100) surface are available for the adsorption of multiple CO2 molecules, we designed a structure containing eight adsorption sites and then optimized the structure with eight CO2 molecules in order to study the mechanism by which multiple CO2 molecules adsorb onto MgAl2O4. As shown in Fig. 4, only three of the eight CO2 molecules were determined according to our calculations to be chemically adsorbed onto the MgAl2O4 surface (black circle). The rest of the CO2 molecules were relatively far from the surface at distances of 3.8–7.8 Å, and interacted with it weakly with average adsorption energy of ∼0.09 eV per molecule. Fig. 4 also shows the charge reorganization of the MgAl2O4 (100) surface and CO2 molecules. In average, 0.54e charge per CO2 molecule was transferred from MgAl2O4 (100) surface to CO2 molecules. This result shows that these five CO2 molecules are physically rather than chemically bound to the surface, as confirmed from their linear molecular shape. The electrostatic repulsion between the CO2 molecules destabilizes the adsorption, but three CO2 molecules are still strongly interacted with the MgAl2O4 surface at the same time. It is worth noting that a considerable number of CO2 molecules can be adsorbed onto MgAl2O4 (100) surface after the chemisorption.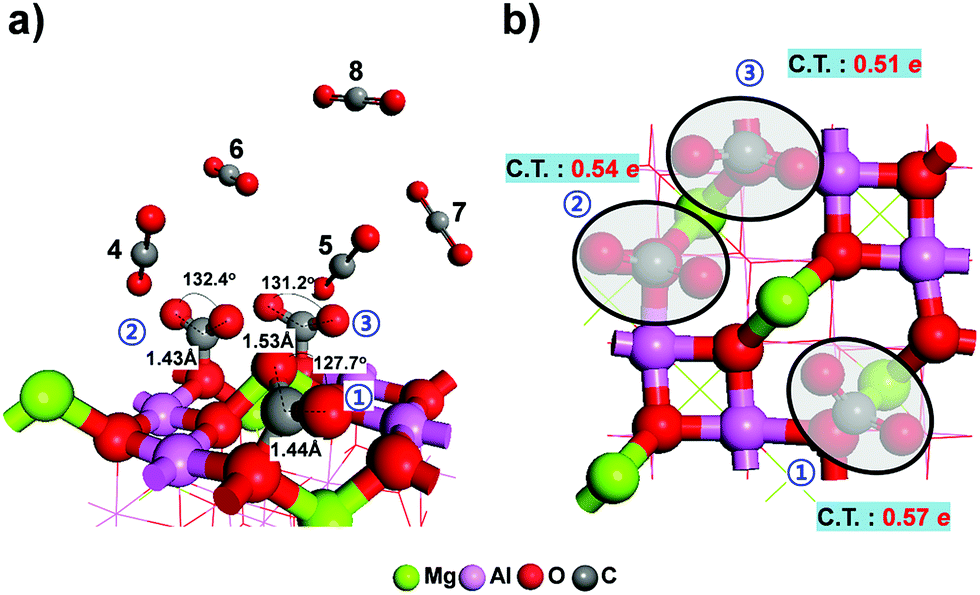 | ||
| Fig. 4 Optimized geometry of the adsorption of multiple CO2 molecules on the MgAl2O4 (100) surface: (a) tilted view, (b) top view. C.T. denotes the charge transfer (e) from the MgAl2O4 (100) surface to CO2 molecules. | ||
It should be noted that the chemisorbed gas molecules cannot be fully reversible and are required high energy for regeneration. For chemisorption, high temperature under constant pressure condition can regenerate the adsorbent for additional reaction of adsorption. In this study, we focused on the evaluation of the inherent properties of the MgAl2O4 (100) surface for CO2 adsorptions without considering the temperature effect using DFT. We will pursue further study on CO2 adsorption and desorption mechanisms with thermal effect.
Conclusions
We investigated CO2 adsorption characteristics on the Mg-based spinel mineral, magnesium aluminate (MgAl2O4) (100), surface by analyzing adsorption energy and configuration using density functional theory (DFT) calculations. The optimized spinel structure of MgAl2O4 is well characterized by DFT calculation with a good agreement with experimental study. It turns out that CO2 molecules were here adsorbed on the sites of two oxygens belonging to octahedral Al3+ and tetrahedral Mg2+ sites with reverse bidentate (with adsorption energy of −1.60 eV) and tridentate (−1.88 eV) bonds, respectively. The MgAl2O4 surface was also found to readily allow both CO2 physisorption and chemisorption for multiple CO2 molecules. This study has also provided a basic understanding of CO2 adsorption mechanism of spinel mineral through computational approach, including procedure and analysis, that may be used to evaluate other spinel group materials such as (Mg, Fe)Al2O4, FeAl2O4, and MnFe2O4.Acknowledgements
This work was supported by KCRC through the NRF funded by Ministry of Science, ICT, and Future Planning (NRF-2015M1A8A1048902). This work was also supported by the Korea Ministry of Environment as Eco-Innovation project (2014000140003).Notes and references
- (a) OECD/IEA, in IEA's Message For Youth on Climate Change, 2014 Search PubMed; (b) C. M. White, B. R. Strazisar, E. J. Granite, J. S. Hoffman and H. W. Pennline, J. Air Waste Manage. Assoc., 2003, 53, 645 CrossRef CAS; (c) R. S. Haszeldine, Science, 2009, 325, 1647 CrossRef CAS PubMed.
- IEA, International Energy Agency (IEA) Statistics Data, 2009 Search PubMed.
- M. G. Plaza, C. Pevida, A. Arenillas, F. Rubiera and J. J. Pis, Fuel, 2007, 86, 2204 CrossRef CAS.
- R. V. Siriwardane and R. W. Stevens, Ind. Eng. Chem. Res., 2009, 48, 2135 CrossRef CAS.
- (a) A. Meisen and X. S. Shuai, Energy Convers. Manage., 1997, 38, S37 CrossRef CAS; (b) R. H. Niswander, D. J. Edwards, M. S. Dupart and J. P. Tse, Sep. Sci. Technol., 1993, 28, 565 CrossRef CAS.
- G. Pacchioni, J. M. Ricart and F. Illas, J. Am. Chem. Soc., 1994, 116, 10152 CrossRef CAS.
- M. M. Maroto-Valer, D. J. Fauth, M. E. Kuchta, Y. Zhang and J. M. Andresen, Fuel Process. Technol., 2005, 86, 1627 CrossRef CAS.
- (a) T. K. Kim, K. J. Lee, J. Y. Cheon, J. H. Lee, S. H. Joo and H. R. Moon, J. Am. Chem. Soc., 2013, 135, 8940 CrossRef CAS PubMed; (b) M. Bhagiyalakshmi, J. Y. Lee and H. T. Jang, Int. J. Greenhouse Gas Control, 2010, 4, 51 CrossRef CAS.
- Y. Bang, S. J. Han, S. Kwon, V. Hiremath, I. K. Song and J. G. Seo, J. Nanosci. Nanotechnol., 2014, 14, 8531 CrossRef CAS PubMed.
- H. J. K. J. G. Seo, S. Kwon and H. C. Lee, in 243rd ACS National Meeting & Exposition, San Diego, California, 2012 Search PubMed.
- (a) M. Tutuianu, O. R. Inderwildi, W. G. Bessler and J. Warnatz, J. Phys. Chem. B, 2006, 110, 17484 CrossRef CAS PubMed; (b) H. Gronbeck, J. Phys. Chem. B, 2006, 110, 11977 CrossRef PubMed.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed; (b) J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- (a) W. Koh, J. H. Lee, S. G. Lee, J. I. Choi and S. S. Jang, RSC Adv., 2015, 5, 32819 RSC; (b) J. H. Lee, S. G. Kang, H. S. Moon, H. Park, I. T. Kim and S. G. Lee, Appl. Surf. Sci., 2015, 351, 193 CrossRef CAS; (c) S. Kwon, D. J. Ham and S. G. Lee, RSC Adv., 2015, 5, 54941 RSC; (d) Y. Yang, Y. Jung, M. D. Cho, S. G. Lee and S. Kwon, RSC Adv., 2015, 5, 57339 RSC; (e) A. Gupta, J. H. Lee, J. H. Seo, S. G. Lee and J. S. Park, RSC Adv., 2015, 5, 73989 RSC; (f) H. Park, S. H. Noh, J. H. Lee, W. J. Lee, J. Y. Jaung, S. G. Lee and T. H. Han, Sci. Rep., 2015, 5, 14163 CrossRef CAS PubMed; (g) J. H. Lee, S. G. Kang, Y. Choe and S. G. Lee, Compos. Sci. Technol., 2016, 126, 9 CrossRef CAS; (h) J. H. Lee, S. G. Kang, I. T. Kim, S. Kwon, I. Lee and S. G. Lee, Theor. Chem. Acc., 2016, 135, 50 CrossRef.
- (a) Q. J. Hong and Z. P. Liu, Surf. Sci., 2010, 604, 1869 CrossRef CAS; (b) S. G. Lee, J. I. Choi, W. Koh and S. S. Jang, Appl. Clay Sci., 2013, 71, 73 CrossRef CAS; (c) Q. L. Tang, Q. J. Hong and Z. P. Liu, J. Catal., 2009, 263, 114 CrossRef CAS; (d) S. Kwon, J. I. Choi, S. G. Lee and S. S. Jang, Comput. Mater. Sci., 2014, 95, 181 CrossRef CAS; (e) S. Kwon, S. G. Lee, E. Chung and W. R. Lee, Bull. Korean Chem. Soc., 2015, 36, 11 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- S. Y. Jing, L. B. Lin, N. K. Huang, J. Zhang and Y. Lu, J. Mater. Sci. Lett., 2000, 19, 225 CrossRef CAS.
- J. P. Schaffer, A. Saxena, S. D. Antolovich, T. H. Sanders and S. B. Warner, The Science and Design of Engineering Materials, 1999 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |