
Role of iron in the reduction of H2O2 intermediate during the oxygen reduction reaction on iron-containing polyimide-based electrocatalysts†
Abstract
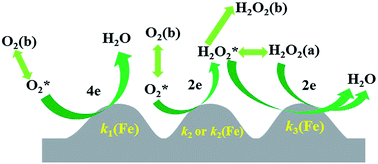
Understanding the role of Fe in each step of the oxygen reduction reaction (ORR) on Fe-containing N-doped carbon catalysts for the ORR in PEMFCs is of great importance in designing higher performance ORR catalysts. The direct 4-electron ORR has been found to be catalyzed by Fe whereas the importance of Fe in the consecutive 2 × 2-electron ORR remains unclear. The reduction of H2O2 on the Fe-containing polyimide (Fe/PI) and Fe-free polyimide (PI) catalysts was studied in acidic media using rotating disk electrode (RDE) voltammetry and the corresponding rate constants were estimated. The results demonstrate that the Fe in the Fe/PI catalyst plays a crucial role in the enhanced H2O2 reduction and consequently a consecutive 2 × 2-electron ORR via adsorbed H2O2 intermediate, i.e., a so-called two-site reduction process, as well as a direct 4-electron ORR could significantly contribute to the overall 4-electron ORR.
 Please wait while we load your content...
Please wait while we load your content...

