
Electronic structure and thermoelectric properties of p-type half-Heusler compound NbFeSb: a first-principles study†
Abstract
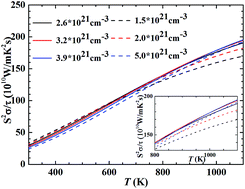
The electronic structure and thermoelectric (TE) properties of p-type NbFeSb are studied by first-principles calculations and the Boltzmann transport equation under the constant relaxation time approximation. The carrier concentration dependences of TE properties of p-type NbFeSb are calculated to be well agreement with the experimental data. When the minimum lattice thermal conductivity is obtained, the maximum ZT of 1.4 is achievable at 800 K, ∼40% higher than the value of 1 for the best p-type Nb1−xHfxFeSb compounds. To further evaluate the optimal electrical transport properties of p-type NbFeSb at higher temperatures, the maximum power factors and corresponding optimal carrier concentrations are calculated. The temperature dependence of Seebeck coefficient and power factor are also studied based on the estimated optimal doping levels, which indicates that further composition optimization can't improve the power factors when the carrier concentration reaches ∼2.6 × 1021 cm−3.
 Please wait while we load your content...
Please wait while we load your content...

