
Energetic stability, STM fingerprints and electronic transport properties of defects in graphene and silicene
Abstract
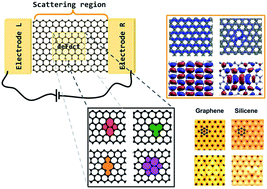
Novel two-dimensional materials such as graphene and silicene have been heralded as possibly revolutionary in future nanoelectronics. High mobilities, and in the case of silicene, its seemingly natural integration with current electronics could make them the materials of next-generation devices. Defects in these systems, however, are unavoidable particularly in large-scale fabrication. Here we combine density functional theory and the non-equilibrium Green's function method to simulate the structural, electronic and transport properties of different defects in graphene and silicene. We show that defects are much more easily formed in silicene, compared to graphene. We also show that, although qualitatively similar, the effects of different defects occur closer to the Dirac point in silicene, and identifying them using scanning tunneling microscopy is more difficult particularly due to buckling. This could be overcome by performing direct source/drain measurements. Finally we show that the presence of defects leads to an increase in local current from which it follows that they not only contribute to scattering, but are also a source of heating.
 Please wait while we load your content...
Please wait while we load your content...

