
Hydrogenation of lignin-derived phenolic compounds over step by step precipitated Ni/SiO2†
Abstract
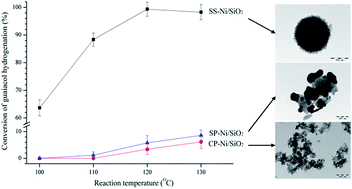
The harsh reaction conditions for the valorization of lignin-derived phenolic compounds considerably limit the efficient utilization of the lignin derivatives. Here, we put forward a high efficient and selective hydrogenation process for phenolic compounds at a mild condition over step by step precipitated Ni/SiO2 catalyst. The properties of the Ni/SiO2 catalysts by different preparation methods were detailedly compared using various characterization measurements. Catalytic activity of the catalysts was tested by the hydrogenation of guaiacol, and the results showed that guaiacol could be completely converted into cyclohexanol with 99.9% selectivity at 120 °C, 2 MPa H2 atmosphere for 2 h. Other typical lignin-derived phenolic compounds also had excellent hydrogenation performance and great energy efficiency. Catalyst characterization results demonstrated that the high catalytic activity of the step by step precipitated Ni/SiO2 was mainly ascribed to its polyporous spherical structure, which led to the large specific surface area and high nickel dispersion. The appropriate acidity of the catalyst also promoted the catalytic performance significantly. Furthermore, the catalyst exhibited an excellent recyclability, where no significant loss of the catalytic activity was showed out after 3 runs.
 Please wait while we load your content...
Please wait while we load your content...

