
Branched polyethyleneimine modified with hyaluronic acid via a PEG spacer for targeted anticancer drug delivery†
Abstract
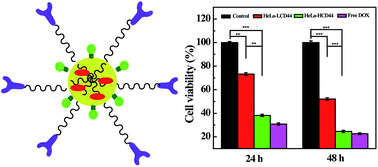
It is generally required to develop a nanocarrier system that is able to improve the water solubility of an anticancer drug and enable targeted delivery of the drug to cancer cells via a receptor-mediated endocytosis pathway. In this work, polyethyleneimine (PEI) was sequentially modified with dual functional polyethylene glycol (NH2–PEG–COOH), hyaluronic acid (HA), and fluorescein isothiocyanate (FI). The prepared PEI–FI–(PEG–HA) conjugate was then used as a nanoplatform to encapsulate the anticancer drug doxorubicin (DOX). We show that the formed PEI–FI–(PEG–HA) conjugate is able to encapsulate approximately 19 DOX molecules within each multifunctional PEI, and the formed PEI–FI–(PEG–HA)/DOX complexes can release DOX in a pH-dependent manner with a higher DOX release rate under an acidic pH condition than under a physiological pH condition. In addition, the PEI–FI–(PEG–HA)/DOX complexes are able to specifically target cancer cells overexpressing CD44 receptors as confirmed via flow cytometric analysis and confocal microscopic observation, and thus deliver DOX to the target cancer cells to inhibit their growth. The developed HA-targeted PEI may hold great promise to be used as an efficient nanoplatform for the targeted delivery of different anticancer drugs.
 Please wait while we load your content...
Please wait while we load your content...

