
Improving photoelectrochemical performance of highly-ordered TiO2 nanotube arrays with cosensitization of PbS and CdS quantum dots†
Abstract
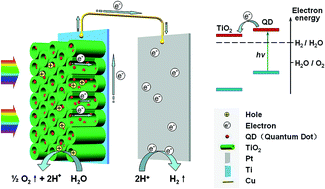
PbS and CdS quantum dots (QDs) were deposited on TiO2 nanotube arrays (TNTAs) by a sonication-assisted successive ionic layer adsorption and reaction (S-SILAR) method. Effects of the composite configuration, QD deposition order, and number of S-SILAR cycles on the photoelectrical properties have been systematically investigated. Depositing the TNTAs with PbS QDs before or after CdS QDs results in TNTAs/PbS/CdS or TNTAs/CdS/PbS configurations. The deposition order has been demonstrated to strongly affect the photoelectrochemical performance, and the TNTAs/PbS/CdS configuration shows the superior performance due to the synergistic effect of the different types of QDs. Studies of the photoelectrical properties of the TNTAs sensitized by single QDs with various number of S-SILAR cycles suggest that PbS decoration can dramatically increase the photocurrent density, and CdS can stabilize the photoelectrochemical behavior. Combining both beneficial effects of these two kinds of QDs, the TNTAs sensitized by 5 cycles PbS and 5 cycles CdS (referred to as TNTAs/PbS(5)/CdS(5)) can achieve the photocurrent density of 0.87 mA cm−2 under a white light source irradiation of 5.9 mW cm−2, corresponding to a record normalized photocurrent of 147 mA W−1. And the photoconversion efficiency can reach 14.3%. Further time-dependent measurement shows that TNTAs/PbS(5)/CdS(5) possesses a stable photoresponse for more than 120 min.
 Please wait while we load your content...
Please wait while we load your content...

