DOI:
10.1039/C5RA22758E
(Paper)
RSC Adv., 2016,
6, 22179-22186
Synthesis of ketoxime-functionalized Fe3O4@C core–shell magnetic microspheres for enhanced uranium(VI) removal
Received
30th October 2015
, Accepted 16th February 2016
First published on 17th February 2016
Abstract
Ketoxime-functionalized carbon coated iron oxide (Fe3O4@C–KO) was synthesized and characterized by transmission electron microscopy, X-ray diffraction, Fourier transformed infrared spectroscopy, and magnetic measurements. The essential factors affecting uranium(VI) adsorption from an aqueous solution of Fe3O4@C–KO, such as initial pH, contact time and temperature, were investigated. The adsorption is highly dependent on solution pH. Analysis of the experimental data using sorption kinetic models, reveal that the process follows a pseudo-second-order kinetic model. In addition, adsorption isotherms and thermodynamics were investigated. The adsorption of uranium(VI) from an aqueous solution onto Fe3O4@C–KO was fitted to Langmuir and Freundlich adsorption isotherms. The adsorption of uranium(VI) is well-described by the Langmuir isotherm. Thermodynamic parameters further show that the sorption is an endothermic and spontaneous process. Fe3O4@C–KO is a powerful and promising sorbent for the efficient removal of uranium(VI) from aqueous solutions.
Introduction
Due to the rapid development of modern industry, heavy metal ions have excessively accumulated in the biosphere and water, leading to a deterioration of the natural environment and a serious health hazard.1,2 Uranium is a typical heavy metal element; it is radioactive and toxic.3–5 Therefore, it is necessary to extract uranium from industrial waste water. Conventional methods for the removal of uranium include chemical precipitation,6 solvent extraction,7 ion-exchange,8 ultrafiltration,9 reverse osmosis and nanofiltration.10 In comparison with these, adsorption techniques have several advantages11–13 including low cost, ease of operation and high efficiency, and have become the predominant methods for the removal and recovery of uranium ions.
In adsorption, as the sorbent is a key factor, much attention has focused on researching new sorbents with fast sorption kinetics, high surface area and good stability for a wide pH range. Traditional sorbents such as oxides and clay material suffer from either low sorption efficiencies or low capacities.
Recently, numerous adsorbents have been developed; these include nanooxides, nanocarbon and carbon-based nanocomposites,14–18 that exhibit excellent sorption capacity. However, the separation process of adsorbents from aqueous solutions after saturated adsorption is usually complex and time-consuming. Magnetic nanoparticles (MNPs), especially Fe3O4, have attracted more attention because of their outstanding properties, such as ease of separation and low toxicity.19–22 Das et al. have studied the sorption of uranium(VI) onto magnetite (Fe3O4) nanoparticles, but the sorption capacity is relatively small. To increase the adsorption capacity, researchers have modified the magnetic nanoparticles with other materials. Ketoxime, an excellent chelate functional group, has been grafted onto the surfaces of various substrates for recovery and removal of uranium(VI) to achieve fast sorption rate, high uranium(VI) loading capacity, and safety of the environment.23–25
Based on a high-throughput, one-pot solvothermal approach, Fe3O4@C nanoparticles (NPs) are synthesized and then modified with ketoxime (KO) on the surface (Fe3O4@C–KO). The prepared Fe3O4@C–KO was used to extract uranium(VI) from aqueous solutions, and the sorption kinetics, effects of geochemical conditions, sorption capability, stability and regeneration were investigated. Based on the experimental findings, the potential application of Fe3O4@C–KO for the recovery and removal of uranium(VI) was evaluated.
Experimental
Material preparation
All the chemical reagents in this work are purchased from Tianjin Kermel Chemical Reagents Development and used without further purification. A solvothermal method was adopted26,27 and slightly modified to control particle size at around 100 nm. Briefly, ferrocene (0.15 g) was dissolved in 30 mL acetone with the aid of ultrasonication for 10 min. 0.5 mL H2O2 was slowly added, and the solution magnetically stirred for 30 min. After that, the precursor solution was transferred into a Teflon-lined stainless autoclave with a capacity of 50 mL. For the reaction, the temperature was maintained at 210 °C for 24 h, before cooling to room temperature naturally. The mixture in the autoclave was subjected to ultrasonic treatment for 10 min. The precipitates were collected by a magnet from the mixture and washed with acetone three times.
Acetophenone (2.00 g) was dissolved in ethanol (55 mL) with stirring. NH2OH·HCl (1.03 g, 1.48 × 10−2 mol) in 20 mL of water and Na2CO3 (0.78 g, 7.40 × 10−3 mol) in 20 mL of water were added to the ethanolic acetophenone solution. The mixture was heated under reflux. With concentration of the solution, yellow, needle-shaped crystals precipitated from solution; these were collected via vacuum filtration and washed with cold water.28
Fe3O4@C powder (0.1 g) was dried at 120 °C overnight and mixed with 0.5 g of acetophenone oxime until a fine black powder was obtained. Then, 4 mL of isoamyl nitrite was added dropwise under vigorous stirring. The stirring was continued for 1 h at room temperature. The reaction mixture was then heated at 70 °C for 2 h before washing five times with dimethylformamide (DMF) and three times with ethanol (EtOH). The resulting black powder was dried under a vacuum.29
Adsorption of uranium(VI)
Uranium removal experiments were performed in a series of conical flasks (100 mL) in which a given dose of the adsorbent was shaken with uranium solution (50 mL), of given concentration and pH, in a thermostatic water shaker at speed of 200 rpm. The pH of the solution was adjusted with 0.5 M HNO3 or NaOH solution. The mixture was shaken for 120 min in a thermostatic shaker bath. After magnetic separation, the concentration of uranium(VI) in the solution was determined by Bruker 820 MS Inductive Coupled Plasma (ICP) instrument. The adsorption capacity Qe (mg g−1) and the % removal of uranium were calculated according to eqn (1) and (2):| |
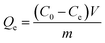 | (1) |
| |
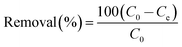 | (2) |
where C0 (mg L−1) is the uranium(VI) ion concentration in the initial solution, Ce (mg L−1) is the equilibrium concentration of uranium(VI) ion in the supernatant, V (L) is the volume of the testing solution and m is the weight of sorbent (g).
Desorption studies
To investigate the reusability of Fe3O4@C–KO, 0.02 g of Fe3O4@C–KO was first put in contact with 20 mL uranium(VI) for 120 min. After adsorption, desorption was carried out by washing the adsorbents with distilled water several times. Then the solution containing 20 mL different concentrations of HCl solution was added into the adsorbed uranium(VI) adsorbents for 120 min. After magnetic separation, the remaining uranium(VI) concentration in the supernatant was measured to evaluate the desorption percentage. The regenerated Fe3O4@C–KO was washed thoroughly with distilled water and then used for the next sorption–desorption cycle.
Characterization
X-ray diffraction (XRD) analysis was performed on a Rigaku D/max-IIIB diffractometer with CuKα irradiation (Kα = 1.54178 Å). The X-ray source was operated at 40 kV and the current used in XRD measurements was 150 mA. Morphology was characterized using transmission electron microscopy (TEM). Powder samples for the TEM observation were dispersed in ethanol by ultrasound and mounted onto a carbon-coated copper microgrid. TEM images were taken by TEM (FEI Tecnai G2 S-Twin) with an acceleration voltage of 200 kV. The magnetic measurement was carried out with a vibrating sample magnetometer (VSM, Lanzhou University LakeShore 7304). Effluent was analyzed using WGJ-III Trace Uranium Analyzer from the Bruker 820-MS ICP instrument.
Results and discussion
Characterization of samples
Fig. 1 shows the XRD patterns of as-synthesized Fe3O4@C–KO. In the XRD pattern of Fe3O4@C–KO (Fig. 1), we see that the diffraction peaks correspond to the (220), (311), (400), (422), (511), (440) and (622) crystalline planes of cubic Fe3O4 (JCPDS75-1609), validating the presence of Fe3O4 nanocrystals within the core–shell structure. The broad band around 22° is ascribed to amorphous carbon,30 which indicates that the core–shell microspheres have been successfully synthesized.
 |
| | Fig. 1 XRD pattern of Fe3O4@C–KO. | |
The magnetic properties of Fe3O4@C–KO were studied at room temperature by measuring the magnetization curves, as shown in Fig. 2. The saturation magnetization (Ms) value is 30.33 emu g−1 for Fe3O4@C–KO. Although the saturation magnetization decreases due to the decrease of the magnetite fraction after carbon coating,31,32 complete magnetic separation is quickly achieved by placing a magnet near the vessels containing t0068e aqueous dispersion of Fe3O4@C–KO particles.
 |
| | Fig. 2 Magnetic hysteresis loop for Fe3O4@C–KO at 300 K. | |
To identify the modification with ketoxime functional groups, FT-IR spectra of Fe3O4@C and Fe3O4@C–KO were recorded (Fig. 3). The peak at 588 cm−1 is attributed to the stretching vibration of the Fe–O bond (Fig. 3 Fe3O4@C). The broad band around 3180 cm−1 corresponds to –OH stretching vibration, and the band around 1720 cm−1 corresponds to the bending vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O,33 reflecting the coating of carbon on the magnetite surface (Fig. 3 Fe3O4@C). After modification with KO (Fig. 3 Fe3O4@C–KO), the CO absorption bands disappear while two new bands arise at 1658 cm−1 and 1010 cm−1 corresponding to the CN and N–O stretching vibrations of ketoxime groups, respectively.34
O,33 reflecting the coating of carbon on the magnetite surface (Fig. 3 Fe3O4@C). After modification with KO (Fig. 3 Fe3O4@C–KO), the CO absorption bands disappear while two new bands arise at 1658 cm−1 and 1010 cm−1 corresponding to the CN and N–O stretching vibrations of ketoxime groups, respectively.34
 |
| | Fig. 3 FT-IR spectra of Fe3O4@C and Fe3O4@C–KO. | |
The morphology of Fe3O4@C and Fe3O4@C–KO were characterized by TEM, which shows in Fig. 4A and B that the obtained Fe3O4@C product is composed of spherical nanoparticles with an average diameter of about 90 nm. As shown in Fig. 4A, the condensation of carbon onto the surfaces of Fe3O4 cores results in microspheres with dark-colored Fe3O4 cores and gray-colored carbon shells having an average thickness of about 30 nm. The clarity of the core–shell structure is due to the distinct density contrast between these two components. The HRTEM image (Fig. 4C) shows a clear lattice between the adjacent fringes. The lattice d-spacing of 0.29 nm corresponding to (220) planes of Fe3O4, is identified in Fig. 4C. The selected area electron diffraction (SAED) pattern (Fig. 4D) obtained from this core–shell structure has a highly symmetrical dotted lattice, which reveals the single-crystalline nature of Fe3O4. To further investigate their microstructure, elemental mapping was employed to investigate the TEM elemental distributions in the unique core–shell structure, as depicted in Fig. 5, which reveals a uniform distribution of Fe, O, C and N.
 |
| | Fig. 4 TEM image of Fe3O4@C ((A)), TEM image, HRTEM image and SAED pattern of Fe3O4@C–KO ((B), (C) and (D)). | |
 |
| | Fig. 5 TEM elemental mappings of Fe3O4@C–KO. | |
Effect of pH on the uranium(VI) adsorption
The pH for the adsorbate solution plays an important role in sorption studies. To study the effect of the uranium(VI) sorption onto Fe3O4@C–KO, a number of batch extraction experiments were carried out in which the pH of the working solution varied from 2 to 12 for 120 min contact time at room temperature. The results in Fig. 6 show that UO22+ adsorption capacity increases with the increase in pH from 2.0 to 6.0. At a pH of 2.0, the adsorption effect is very weak because of effective competition between high concentrations of H+ and H3O+.35
 |
| | Fig. 6 Effect of initial pH on adsorption of uranium by Fe3O4@C–KO. (Adsorption dosage 0.02 g, retention time 120 min, T = 25 °C and pH = 2–12). | |
The adsorption capacity reaches a maximum at pH 6.0 and diminishes as the pH rises from 6.0 to 12.0. With a pH higher than 6.0, uranium is present in an anionic form by complexation with carbonate and hydroxyl anions36,37 which has less interaction with functional groups of Fe3O4@C–KO leading to a decrease in adsorption. Consequently, pH 6.0 is considered as the optimum pH for further experiments.
Effect of contact time on uranium sorption
Since the contact time between the adsorbate and adsorbent is a key parameter for the adsorption process, the effect of contact time on adsorption of uranium(VI) onto Fe3O4@C–KO was investigated to determine the equilibrium point. The adsorption experiments were carried out for contact times ranging from 15 to 480 min, with 20 mg adsorbents and 20 mL of 50 mg L−1 uranium(VI) solution at 25 °C, and with an initial pH of 6.0 for Fe3O4@C–KO. We observe that uranium(VI) uptake increases sharply in the first 60 min, and does not change significantly after 120 min (Fig. 7). Uranium(VI) adsorption achieves an equilibrium around 120 min. We interpret from the initial rapid adsorption of uranium(VI) that Fe3O4@C–KO has a high specific surface area and relatively large pore sizes; as a result, a shaking time of 120 min is appropriate for maximum sorption of uranium(VI) onto Fe3O4@C–KO.
 |
| | Fig. 7 Effect of reaction time on the adsorption of uranium by Fe3O4@C–KO. (Adsorption dosage 0.02 g, reaction time 5–300 min, T = 25–55 °C and pH = 6). | |
Adsorption kinetics
To investigate the kinetic mechanism, which controls the adsorption process, the kinetics of uranium(VI) adsorption was modeled using pseudo-first-order and second-order rate equations. The first-order equation is written as:| |
ln(qe − qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − k1t qe − k1t
| (3) |
where k1 is the rate constant of pseudo-first-order adsorption, qe and qt (mg g−1) is the amount of uranium adsorbed at equilibrium and at time (t), respectively.
And the pseudo-second-order equation38 is given as:
| | |
t/qt = 1/k2qe2 + t/qe
| (4) |
where
k2 is the rate constant of pseudo-second-order adsorption. By plotting ln(
qe −
qt)
versus t, the values of
k1 and
qe in pseudo-first-order equation are obtained from the slope and intercept (
Fig. 8A), and by the same method,
k2 and
qe in pseudo-second-order equation are obtained (
Fig. 8B). The calculated kinetic parameters from both model fittings are shown in
Table 1.
 |
| | Fig. 8 Pseudo-first-order kinetics and pseudo-second-order kinetics for removal of uranium by Fe3O4@C–KO. | |
Table 1 Kinetic parameters for adsorption of uranium on Fe3O4@C–KO
| Kinetic model |
T (oC) |
C0 (mg L−1) |
Qexpe (mg g−1) |
Qcale (mg g−1) |
k1 (min−1)/k2 (g mg−1 min−1) |
R2 |
| Pseudo-first order |
25 |
60 |
27 |
3.34 |
0.0051 |
0.6893 |
| Pseudo-second order |
25 |
60 |
27 |
26.88 |
0.0076 |
0.9998 |
In pseudo-second-order kinetics, the calculated qe values are nearly the same as the experimental values, and the regression coefficient is 0.9998, which confirms that the adsorption of uranium(VI) ions adsorption onto Fe3O4@C–KO is described by a pseudo-second-order model. These results suggest that a pseudo-second-order adsorption is the predominant mechanism and the rate constant of uranium(VI) ions appears to be controlled by a chemisorption process.
Effect of temperature and adsorption thermodynamics
The effect of temperature on uranium(VI) sorption onto Fe3O4@C–KO was investigated using a water bath with 20 mg adsorbent, 20 mL of uranium(VI) 50 mg L−1, 120 min contact time, and pH of 6.0 for Fe3O4@C–KO. The sorption of uranium(VI) onto Fe3O4@C–KO is highest at 328 K the lowest at 298 K (Fig. 9), indicating that high temperature is advantageous for uranium(VI) sorption.
 |
| | Fig. 9 Effect of uranium concentration on the adsorption of uranium by Fe3O4@C–KO. (Adsorption dosage 0.02 g, reaction time 120 min, T = 25–55 °C and pH = 6). | |
Entropy (ΔS°) and enthalpy (ΔH°) changes, were calculated using the following equation:39
| | |
lnKd = −ΔH°/RT + ΔS°/R
| (5) |
where
Kd is the equilibrium constant (mL g
−1), Δ
H° is standard enthalpy (kJ mol
−1), Δ
S° is standard entropy (J mol
−1 K
−1),
T is the absolute temperature (K), and
R is the gas constant (8.314 J mol
−1 K
−1). The values of Δ
H° and Δ
S° are evaluated from the intercept and slope of the linear plot of ln
Kd vs. 1/
T (
Fig. 10). The positive standard entropy (Δ
S°) means that randomness is increases at the solid/solution interface during adsorption. The positive value of Δ
H° indicates the adsorption process of UO
22+ is endothermic.
 |
| | Fig. 10 Van't Hoff plot for removal of uranium by Fe3O4@C–KO. | |
The thermodynamic parameter, ΔG°, is calculated from the following Gibbs–Helmholtz equation:
where Δ
G° is the standard Gibbs free energy. From
eqn (6), the data of Δ
G° at different temperatures are obtained. The data of Δ
G°, Δ
H° and Δ
S° are shown in
Table 2. The negative values of Δ
G° indicate that the adsorption follows a spontaneous and feasible trend. Gibbs free energy decreases with increase in temperature, which suggests that higher temperatures facilitate adsorption of uranium(
VI) ions onto Fe
3O
4@C–KO due to a greater driving force.
Table 2 Thermodynamic parameters for adsorption of uranium on Fe3O4@C–KO
| ΔH° (kJ mol−1) |
ΔS° (J mol−1 K−1) |
ΔG° (kJ mol−1) |
| 10.21 |
40.43 |
298 K |
308 K |
318 K |
328 K |
| −1.76 |
−2.24 |
−2.65 |
−3.05 |
Adsorption isotherms
For the design and operation of adsorption systems, correlation of equilibrium adsorption data is important. Langmuir and Freundlich isotherms models were applied to the obtained adsorption data.
The Langmuir equation has been used extensively for dilute solutions in the following form:40
| | |
Ce/qe = 1/bqm + Ce/qm
| (7) |
where
Ce (mg L
−1) is the equilibrium concentration of UO
22+ remained in solution,
qe (mg g
−1) is the amount of solution adsorbed per unit mass of the adsorbent,
qm (mg g
−1) is the maximum adsorption capacity,
b is the Langmuir adsorption equilibrium constant. According to
eqn (7), a straight line is obtained and presented in
Fig. 11A. The values of
qm and
b are calculated from the slope and the intercept, and are given in
Table 3.
 |
| | Fig. 11 Langmuir and Freundlich isotherm for removal of uranium by Fe3O4@C–KO. | |
Table 3 Langmuir and Freundlich isotherm parameters for adsorption of uranium on Fe3O4@C–KO
| Parameter |
Value |
R2 |
| Langmuir isotherm Qm (mg g−1) |
38.76 |
0.9567 |
| b (L mg−1) |
0.0472 |
|
| Freundlich isotherm K (L g−1) |
3.578 |
0.9222 |
| n |
1.866 |
— |
The Freundlich model is based on a reversible heterogeneous adsorption since it is not restricted to monolayer adsorption capacity.38
The Freundlich isotherm is given as:
where
k and
n are the Freundlich constants related to the adsorption capacity and adsorption intensity, respectively. They are determined from the intercept and slope of the linear plot of ln
qe vs. ln
Ce (
Fig. 11B).
The corresponding Langmuir and Freundlich parameters, along with the correlation coefficients, are reported in Table 3.
Table 3 shows that the Langmuir isotherm model better fits the experimental results over the experimental range, with high correlation coefficients (>0.95). The Langmuir model indicates that it is monolayer adsorption onto structurally homogeneous Fe3O4@C–KO. The maximum adsorption capacity of Fe3O4@C–KO is evaluated as 38.76 mg U g−1 at 25 °C.
Desorption and reusability study
To test the feasibility of Fe3O4@C–KO materials to be regenerated after adsorption of uranium(VI) ions, desorption was carried out with Fe3O4@C–KO adsorbent. Uranium(VI)-loaded adsorbent was regenerated using HCl solution. Various concentrations of HCl ranging between 0.01 and 1.0 M were tested for elution of adsorbed uranium(VI) ions using Fe3O4@C–KO. The percentage desorption of HCl concentrations of 0.01, 0.05, 0.1, 0.5 and 1.0 M is 68.0%, 75.0%, 78.0%, 87.0%, 87.1%, respectively. Therefore, the best and optimum concentration of HCl is determined as 0.5 M in terms of economic efficiency.
To assess the reusability of the adsorbent, the adsorption–desorption experiment with 0.5 M HCl was repeated for three cycles. After three cycles, the desorption efficiency of the Fe3O4@C–KO is still more than 80%. This result shows that the adsorbent can be used efficiently in a real process such as nuclear industry wastewater treatment.
Comparison of adsorbent performance with literature data
The removal of uranium(VI) by different adsorbents has been studied extensively. Table 4 represents the comparison of the adsorption capacity of uranium(VI) with other materials.41–45 Adsorption capacity of Fe3O4@C–KO equal to 38.76 mg U g−1 is higher than that of other adsorbents, except for oxime-grafted CMK-5. This data suggests that the Fe3O4@C–KO as adsorbent is suitable for the removal of uranium(VI) from aqueous solution.
Table 4 Comparison of the uranium(VI) sorption capacity of Fe3O4@C–KO with other sorbents
| Sorbents |
Capacity (mg U g−1) |
Ref. |
| Hematite |
3.36 |
41 |
| Functionalized polymer-coated silica |
5.2 |
42 |
| Amine modified silica gel |
21.4 |
43 |
| Activated carbon |
28.3 |
44 |
| Oxime-grafted CMK-5 |
62 |
45 |
| Fe3O4@C–KO |
38.7 |
Present work |
Conclusions
A novel magnetic Fe3O4@C–KO composite has been fabricated, with its structure well-characterized by FT-IR, XRD, TEM and VSM. The adsorption process is pH dependent and accomplished within 120 min. The adsorption kinetic process is well-described by a pseudo-second-order model. Thermodynamic studies indicate an endothermic and spontaneous adsorption process. In addition, uranium(VI)-loaded Fe3O4@C–KO is easily separated from aqueous solutions by a magnet and efficiently renewed by HCl. The ease of operation and fast and efficient adsorption performance indicate that Fe3O4@C–KO can be used as a highly effective material for the removal of uranium(VI) ions from aqueous solution.
Acknowledgements
This work was supported by National Natural Science Foundation of China (51402065), Heilongjiang Province Natural Science Funds for Distinguished Young Scholar (JC201404), Special Innovation Talents of Harbin Science and Technology for Distinguished Young Scholar (2014RFYXJ005), Fundamental Research Funds of the Central University (HEUCFZ), Natural Science Foundation of Heilongjiang Province (B201404), Program of International S&T Cooperation special project (2015DFR50050), Special Innovation Talents of Harbin Science and Technology (2013RFQXJ145, 2014RFQXJ013), and the fund for Transformation of Scientific and Technological Achievements of Harbin (2013DB4BG011), Natural Science Foundation of Heilongjiang Province (E201329), Innovation Talents of Harbin Science and Technology (2014RFQXJ035).
Notes and references
- S. J. DeCanio and A. Fremstad, Energy Policy, 2011, 39, 1144–1153 CrossRef CAS.
- B. Volesky and Z. R. Holan, Biotechnol. Prog., 1995, 11, 235–250 CrossRef CAS PubMed.
- H. Nifenecker, Future electricity production methods. Part 1: nuclear energy, Rep. Prog. Phys., 2011, 74(2), 1–28 CrossRef.
- M. Asif and T. Muneer, Renewable Sustainable Energy Rev., 2007, 11(7), 1388–1413 CrossRef.
- Y. Lu, Nat. Chem., 2014, 6, 175–177 CrossRef CAS PubMed.
- A. E. Santos and A. C. Q. Ladeira, Environ. Sci. Technol., 2011, 45, 3591–3597 CrossRef PubMed.
- M. Koh, J. Yoo, Y. Park, D. Bae, K. Park, H. Kim and H. Kim, Ind. Eng. Chem. Res., 2006, 45, 5308–5313 CrossRef CAS.
- S. Y. Bae, G. L. Southard and G. M. Murray, Anal. Chim. Acta, 1999, 397, 173–181 CrossRef CAS.
- A. J. C. Semiao, H. M. A. Rossiter and A. I. Schafer, J. Membr. Sci., 2010, 348, 174–180 CrossRef CAS.
- M. G. Khedr, Desalination, 2013, 321, 47–54 CrossRef.
- M. Hua, S. Zhang, B. Pan, W. Zhang, L. Lv and Q. Zhang, J. Hazard. Mater., 2012, 317, 211–212 Search PubMed.
- Y. Ni, L. Jin, L. Zhang and J. Hong, J. Mater. Chem., 2010, 20, 6430–6436 RSC.
- A. Tripathi, J. S. Melo and S. F. D'Souza, J. Hazard. Mater., 2013, 246, 87–95 CrossRef PubMed.
- G. X. Zhao, L. Jiang, Y. D. He, J. X. Li, H. L. Dong, X. K. Wang and W. P. Hu, Adv. Mater., 2011, 23, 3959–3963 CrossRef CAS PubMed.
- Y. B. Sun, S. T. Yang, G. D. Sheng, Z. Q. Guo, X. L. Tan, J. Z. Xu and X. K. Wang, Sep. Purif. Technol., 2011, 83, 196–203 CrossRef.
- D. D. Shao, Z. Q. Jiang, X. K. Wang, J. X. Li and Y. D. Meng, J. Phys. Chem. B, 2009, 113, 860–864 CrossRef CAS PubMed.
- H. Chen, J. X. Li, D. D. Shao, X. M. Ren and X. K. Wang, Chem. Eng. J., 2012, 210, 475–481 CrossRef CAS.
- J. X. Li, S. Y. Chen, G. D. Sheng, J. Hu, X. L. Tan and X. K. Wang, Chem. Eng. J., 2011, 166, 551–558 CrossRef CAS.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed.
- I. Akin, G. Arslan, A. Tor, M. Ersoz and Y. Cengeloglu, J. Hazard. Mater., 2012, 235, 62–68 CrossRef PubMed.
- H. Y. Koo, H. J. Lee, H. A. Go, Y. B. Lee, T. S. Bae, J. K. Kim and W. S. Choi, Chem.–Eur. J., 2011, 17, 1214–1219 CrossRef CAS PubMed.
- J. X. Li, Z. Q. Guo, S. W. Zhang and X. K. Wang, Chem. Eng. J., 2011, 172, 892–897 CrossRef CAS.
- S. Das, A. K. Pandey, A. A. Athawale and V. K. Manchanda, J. Phys. Chem. B, 2009, 113, 6328–6335 CrossRef CAS PubMed.
- O. Hazer and S. Kartal, Talanta, 2010, 82, 1974–1979 CrossRef CAS PubMed.
- A. Y. Zhang, G. Uchiyama and T. Asakura, React. Funct. Polym., 2005, 63, 143–153 CrossRef CAS.
- Q. An, M. Yu, Y. T. Zhang, W. F. Ma, J. Guo and C. C. Wang, J. Phys. Chem. C, 2012, 116, 22432–22440 CAS.
- Q. An, P. Zhang, J. M. Li, W. F. Ma, J. Guo, J. Hu and C. C. Wang, Nanoscale, 2012, 4, 5210–5216 RSC.
- B. Christer, Y. Aakero, M. Alicia and S. Destin, Cryst. Growth Des., 2001, 1, 47–52 Search PubMed.
- M. Carboni, C. W. Abney, M. L. Kathryn, L. Juan, V. Escoto and W. B. Lin, Ind. Eng. Chem. Res., 2013, 52, 15187–15197 CrossRef CAS.
- U. Kalapathy, A. Proctor and J. Shultz, Bioresour. Technol., 2000, 73, 257–262 CrossRef CAS.
- J. H. Wang, S. R. Zheng, Y. Shao, J. L. Liu, Z. Y. Xu and D. Q. Zhu, J. Colloid Interface Sci., 2010, 349, 293–299 CrossRef CAS PubMed.
- S. H. Xuan, L. Y. Hao, W. Q. Jiang, X. L. Gong, Y. Hu and Z. Y. Chen, Nanotechnology, 2007, 18, 035602 CrossRef PubMed.
- S. Sadeghi, H. Azhdari, H. Arabi and A. Z. Moghaddam, J. Hazard. Mater., 2012, 215, 208–216 CrossRef PubMed.
- B. J. Gao, Y. C. Gao and Y. B. Li, Chem. Eng. J., 2010, 158, 542–549 CrossRef CAS.
- A. Schierz and H. Zaker, Environ. Pollut., 2009, 157, 1088–1094 CrossRef CAS PubMed.
- M. Sutton and S. R. Burastero, Chem. Res. Toxicol., 2004, 17, 1468–1480 CrossRef CAS PubMed.
- M. Wazne, G. P. Korfiatis and X. G. Meng, Environ. Sci. Technol., 2003, 37, 3619–3624 CrossRef CAS PubMed.
- Y. S. Ho and G. McKay, Water Res., 2000, 34, 735–742 CrossRef CAS.
- G. Moussavi and R. Khosravi, Chem. Eng. Res. Des., 2011, 89, 2182–2189 CrossRef CAS.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- A. Mellah, S. Chegrouche and M. Barkat, J. Colloid Interface Sci., 2006, 296, 434–441 CrossRef CAS PubMed.
- D. E. Bryant, D. I. Stewart, T. P. Kee and C. S. Barton, Environ. Sci. Technol., 2003, 37, 4011–4016 CrossRef CAS PubMed.
- K. A. Venkatesan, V. Sukumaran, M. P. Antony and P. R. V. Rao, J. Radioanal. Nucl. Chem., 2004, 260, 443–450 CrossRef CAS.
- S. B. Xie, C. Zhang, X. H. Zhou, J. Yang, X. J. Zhang and J. S. Wang, J. Environ. Radioact., 2009, 100, 162–166 CrossRef CAS PubMed.
- G. Tian, J. X. Geng, Y. D. Jin, C. L. Wang, S. Q. Li, Z. Chen, H. Wang, Y. S. Zhao and S. J. Li, J. Hazard. Mater., 2011, 190, 442–450 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 












