
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and optical properties of covalently bound Nile Red in mesoporous silica hybrids – comparison of dye distribution of materials prepared by facile grafting and by co-condensation routes†
Markus
Börgardts
a,
Kathrin
Verlinden
b,
Manuel
Neidhardt
c,
Tobias
Wöhrle
c,
Annika
Herbst
b,
Sabine
Laschat
c,
Christoph
Janiak
b and
Thomas J. J.
Müller
*a
aInstitut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: ThomasJJ.Mueller@uniduesseldorf.de
bInstitut für Anorganische Chemie und Strukturchemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany
cInstitut für Organische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
First published on 22nd December 2015
Abstract
The fluorescence dye Nile Red (NR) can be covalently ligated to hexagonally ordered, mesoporous silica materials (MCM-41) via co-condensation and post grafting routes in order to investigate possible differences in the dye distributions. The obtained hybrid materials display emission properties similar to free NR but in contrast to free dye molecules even aqueous gels are luminescent, rendering these materials particularly interesting for biolabeling applications. All materials were structurally characterized by nitrogen sorption measurements, small angle X-ray scattering (SAXS), and transmission electron microscopy (TEM). Their optical properties were characterized by UV/Vis and fluorescence spectroscopy.
Introduction
After the discovery of pure inorganic mesoporous silica materials in 1992, research on mesoporous composite materials modified by organic molecules quickly evolved.1–3 By chemical and physical manipulation of their pore systems, the properties of rigid silica and various functional molecules can be combined and lead to unique features of the hybrid materials in comparison to the individual components. As a consequence these novel composite materials open novel applications in various topical fields of research, such as catalysis,4 optical sensing,5,6 solid state lasers,7 and drug delivery.8–10 For sensing applications mesoporous silica hybrids are particularly advantageous due to their pore structure (e.g. MCM-41) enabling facile mass transport at high rates, which is essential for quick response times to environmental changes. Furthermore, the shape and size of silica particles can be controlled by varying the synthetic conditions, thus rendering them also favorable for medicinal applications, such as drug delivery or biolabeling.11 Special sensitivity can be additionally introduced by modification of the silica surface with biochemical functionalities, such as antibodies.9The simplest route of doping organic dyes into sol–gel matrices is the adsorption and entrapping of the chromophores during their synthesis. However, plain adsorption inevitably causes leakage and migration of the dyes, although this problem can be circumvented by covalent ligation of the dye to free silanol groups in the silica framework. In this manner, a dye containing a reactive functionality can be grafted onto the silanol groups of the silica matrix in the sense of a postsynthetic functionalization. Alternatively and quite elegantly in a highly convergent fashion, a dye with pending trialkoxysilyl functionality can be covalently anchored in the mesoporous structure in statu nascendi (co-condensation).12 Yet, the determination of the homogeneity of the dye distribution, especially at low dye loading proved to be difficult and only little research has been dedicated to the comparison of dye distribution of hybrid materials synthesized by post grafting methods and one-pot synthesis. Especially there is barely any comparison between the two most feasible synthesis methods of hybrid materials with similar structural properties, i.e. grafting onto commercially available mesoporous silica and co-condensation.13–15
Taking advantage of the inherent chemical, thermal and dimensional stability of silica hybrid materials various fluorescent dyes have already been incorporated into mesoporous silica by adsorption or covalent ligation, thus enhancing the photostability of the organic components.16,17 Most advantageously a co-condensation strategy could give rise to a homogenous distribution, thereby avoiding aggregation-induced self-quenching of the dye molecules.18,19
A typical fluorescence dye displaying emission self-quenching in the solid state is Nile Red (NR). NR possesses high fluorescence quantum yields in lipids and unpolar solvents and is widely used in biological applications as a lipophilic stain or a laser dye.20–22 Moreover, the pronounced emission solvatochromicity allows its use as polarity sensor in cellular environments.23,24 Unfortunately, NR's water insolubility restricts its application to lipids and highly hydrophobic micro-environments. But still since NR's fluorescence does not interfere with the cellular autofluorescence (typically below 550 nm), NR could be an ideal probe for biolabeling and thus recent work is dedicated to water-soluble, but still luminescent NR derivatives.25–27 As a consequence a new class of luminescent stains for intracellular imaging could evolve.
Upon incorporation of NR into mesoporous silica, aggregation-induced self-quenching in aqueous media should be suppressed, leading to hybrid materials suitable for biotechnological applications.28,29 In addition, these hybrids could be ligated to biomolecules by decorative functionalization of the outer surface of silica particles.9
Finally, these NR-silica hybrids should be still luminescent in the solid sol state. Potential applications can be envisioned in solid state dye lasers where the luminescent properties of the dye are concatenated with the inherent stability of the silica host structure, concomitantly avoiding toxic solvents and ensuring easier handling with potentially adjustable shape, e.g. as fibres.7,30 Moreover, solid-state fluorescent NR hybrids could be employed in security technology as near-infrared solid state dyes.31
Here we report the synthesis of NR-functionalized hybrid silica by the two most feasible synthetic methods i.e. postsynthetic grafting of commercial available MCM-41 and in situ co-condensation. Materials prepared by both routes were studied with respect to homogeneity as well as the effect of applied synthesis route and dye loading of the hybrid. Furthermore, the structural and optical properties of these novel hybrid materials are thoroughly studied and discussed.
Results and discussion
Synthesis
For the covalent ligation of NR to the silica materials a side chain with a terminal triethoxysilyl group had to be introduced. Starting from 3-diethylaminophenol (1), via the formation of the nitroso derivative 2, the 2-hydroxy substituted NR derivative 3 was obtained according to a literature procedure (Scheme 1).32 By Williamson ether synthesis the phenol 3 was transformed into the propargyl ether 4 in 24% yield. Finally, by CuAAC (Cu-catalyzed alkyne-azide cycloaddition)33 of alkyne 4 with (3-azidopropyl)triethoxysilane the required terminal triethoxysilyl group was introduced34 to give the triethoxysilyl functionalized NR precursor 5 in 45% yield.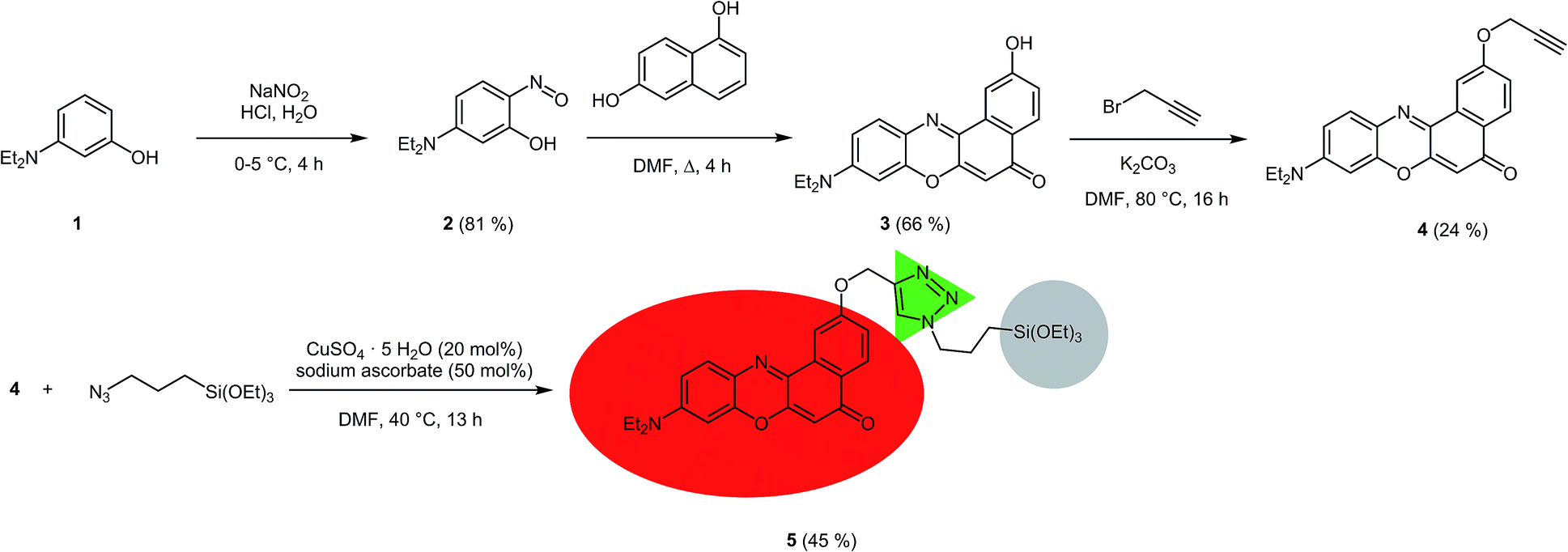 | ||
| Scheme 1 Synthesis of the triethoxysilyl functionalized NR precursor 5. | ||
The NR silica hybrid materials 6 were synthesized via postsynthetic functionalization of commercially available MCM-41 with the precursor molecule 5 (postsynthetic grafting) and NR silica hybrid materials 7 were prepared via in situ co-condensation upon simultaneous formation of the mesoporous structure (in situ synthesis). To exclude dye leakage from the hybrid materials, Soxhlet extraction was performed until no more dye could be detected in the supernatant.
During the postsynthetic grafting, the triethoxysilane terminus of precursor 5 reacts with the free silanol groups exposed on the surfaces in the pores. The major advantage of this strategy is retaining the initial pore structure of the silica material. However, the major drawback is the potential non-uniform distribution of the organic molecules and pore clogging at elevated substrate concentrations as a consequence of preferential functionalization at the pore openings.
In contrast, a homogenous distribution can be achieved by the in situ co-condensation approach where tetraethoxysilane (TEOS), the precursor dye molecule, and the templating agent are present in the same pot. Especially in cases where self-quenching of the emission of fluorophores can be expected at higher concentrations a homogeneous distribution and dilution is indispensable. Furthermore, it is still possible to obtain materials at higher degrees of loading since pore clogging is not an issue. However, since the dye molecules are immediate components of the silica material, an increase in organic functionalization might cause a decrease in mesoporous order, inevitably causing a total collapse of the structural order at high doping levels (Scheme 2).
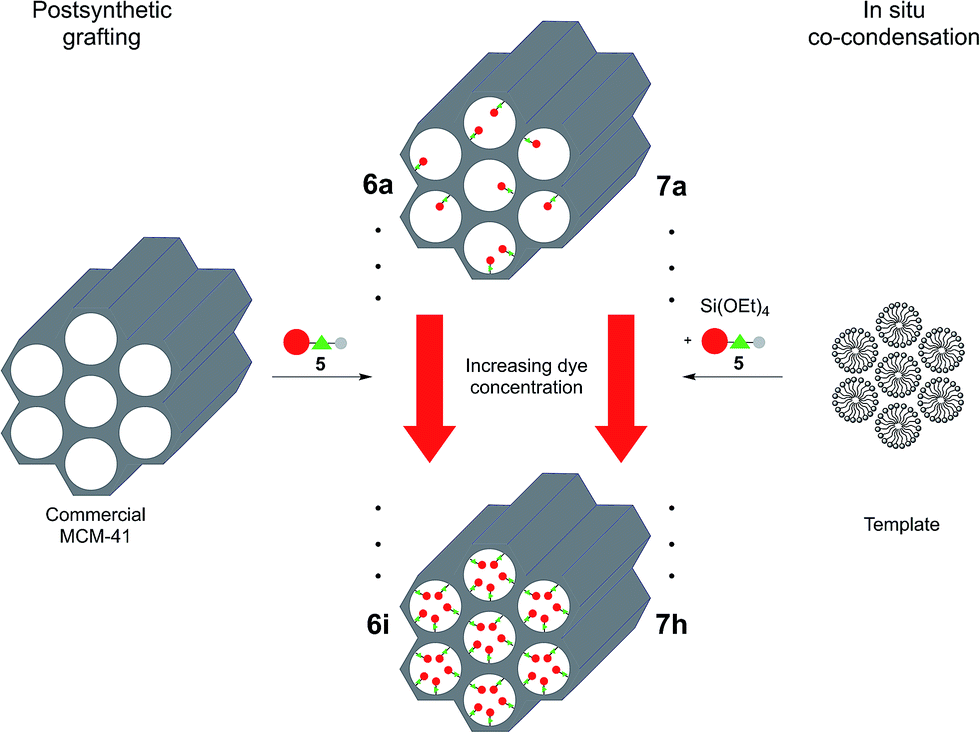 | ||
| Scheme 2 Synthesis of grafted (6a–i) and co-condensed (7a–h) hybrid materials with different dye loadings. | ||
Structural characterization
 | ||
| Fig. 1 N2-sorption isotherms of a grafted (6i), a co-condensed (7h) hybrid material and pure MCM-41. See Fig. S1 and S2 in ESI† for isotherms of 6a–h and 7a–g. | ||
A further distinction between the co-condensed (7) and grafted (6) materials can be made by means of their specific surface areas of ∼1000 and ∼800 m2 g−1, respectively (Table 1). These differences can be rationalized by the modified synthesis conditions of the co-condensed material relative to the synthesis of pure MCM-41 material. Within the series of co-condensed materials, hybrids 7f–h exhibit slightly lower surface areas and pore volumes than the other samples, as well as higher structural disorder, as can be seen in the hysteresis loop of the capillary condensation step. This could be due to a higher dye content inside the pores as this would cause a decrease in surface area. But it is more likely that this decrease is dependent on the modified synthesis conditions as it is known that the structure of MCM materials can be controlled by the addition of alcohols.40–42 Thus, as in the synthesis of 7f–h a higher amount of methanol was applied due to solubility issues of the precursor molecule, it is very likely that this caused a decrease in the surface area rather than a higher dye content in the micromolar range. For the grafted hybrids 6a–i the surface areas range between 748 and 878 m2 g−1, whereas neat MCM-41 has a surface area of 806 m2 g−1. As the experimental error in BET surface area determination can be ±50 m2 g−1 the values overlap with their error margins. So functionalized materials 6a–i do not differ much from neat silica, indicating that dye doping with 0.6–23 μmol g−1 does not lead to a significant variation of the surface area and pore volume.
The pore size distributions of the hybrid materials were obtained by DFT calculations from the N2 isotherms (Fig. 2, S3 and S4 in ESI†). These calculated distributions are bimodal and relatively narrow around diameters of ca. 1.5 and 2.8 nm for MCM-41 and its co-condensed and grafted hybrids. Noteworthy, the volume fraction of the smaller pores around 1.5 nm and of pores larger than 3 nm has decreased in 6 and especially 7 compared to neat MCM-41. As there is no decrease of the volume fraction for low and high loaded materials, this change is attributed to the dye extraction conditions leading to slightly modified pores.
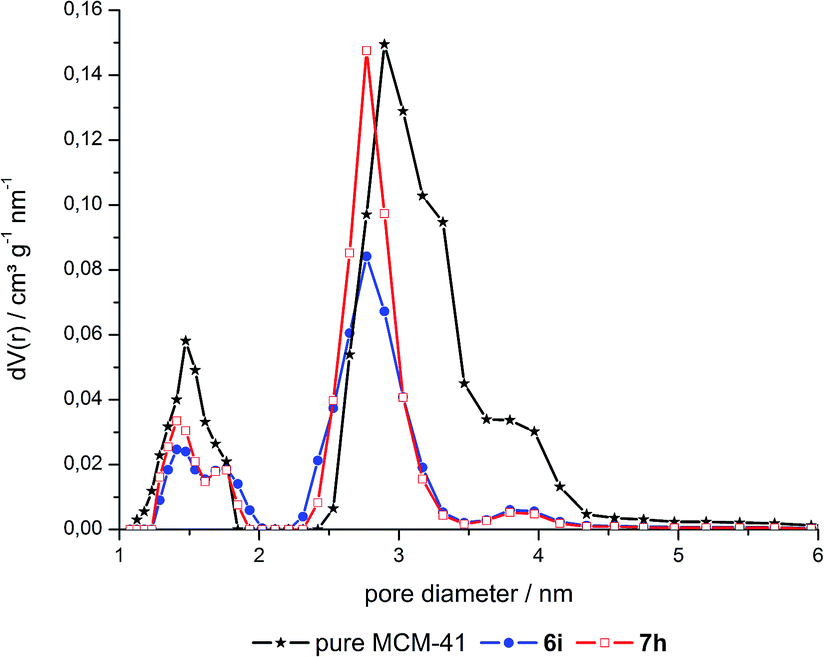 | ||
| Fig. 2 Pore size distribution of a grafted hybrid (6i), a co-condensed hybrid (7h) and pure MCM-41. | ||
| Grafted | Co-condensed | ||||
|---|---|---|---|---|---|
| Sample | Pore volume [cm3 g−1] | BET surface area [m2 g−1] | Sample | Pore volume [cm3 g−1] | BET surface area [m2 g−1] |
| Neat MCM-41 | 0.77 | 806 | |||
| 6a | 0.66 | 808 | 7a | 0.93 | 1042 |
| 6b | 0.69 | 868 | 7b | 0.95 | 1060 |
| 6c | 0.65 | 748 | 7c | 0.93 | 1044 |
| 6d | 0.71 | 878 | 7d | 0.94 | 1061 |
| 6e | 0.64 | 781 | 7e | 0.97 | 1075 |
| 6f | 0.63 | 750 | 7f | 0.81 | 995 |
| 6g | 0.69 | 851 | 7g | 0.77 | 995 |
| 6h | 0.66 | 821 | 7h | 0.74 | 944 |
| 6i | 0.66 | 852 | |||
These results imply that the surface area as well as the pore size distribution is an inherent feature of the employed silica material or rather its synthesis conditions. They are not affected by the loading with varying amounts of dye. The shape of the sorption isotherms and their capillary condensation steps show no distinct change or shift at higher loadings of dye as was the case for organic molecules at concentrations up to 0.77 mmol g−1 incorporated in the MCM-41 type material.43
Small angle X-ray scattering (SAXS)
In order to gain deeper insight into the structure of the hybrid materials the samples were studied by small angle X-ray scattering (SAXS) (Table 2, Fig. 3). The post-grafted hybrid materials 6a–i exhibit 4 reflections corresponding to the (100), (110), (200) and (210) planes, whose 1/d values match with a relation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :√3:√4:√7 which is typical for two-dimensional ordered hexagonal mesostructured material possessing P6mm symmetry.38,44 Thus, no change in the mesostructure can be observed in comparison to neat MCM-41. Furthermore, the post-grafted hybrids as well as the neat MCM-41 exhibit lattice planes around 4.0 nm which is almost unchanged compared to the co-condensed hybrid materials. Like in the gas sorption analysis, the co-condensed materials 7g–h show slightly smaller lattice planes as a consequence of the adjusted synthesis conditions.
:√3:√4:√7 which is typical for two-dimensional ordered hexagonal mesostructured material possessing P6mm symmetry.38,44 Thus, no change in the mesostructure can be observed in comparison to neat MCM-41. Furthermore, the post-grafted hybrids as well as the neat MCM-41 exhibit lattice planes around 4.0 nm which is almost unchanged compared to the co-condensed hybrid materials. Like in the gas sorption analysis, the co-condensed materials 7g–h show slightly smaller lattice planes as a consequence of the adjusted synthesis conditions.
| Grafted | Co-condensed | ||||||
|---|---|---|---|---|---|---|---|
| Sample | b(5)a [μmol g−1] | a [nm] | d 100 [nm] | Sample | b(5)a [μmol g−1] | a [nm] | d 100 [nm] |
| a Loading of hybrid with 5. | |||||||
| 6a | 0.6 | 4.71 | 4.08 | 7a | 0.04 | 4.65 | 4.03 |
| 6b | 1.5 | 4.61 | 4.00 | 7b | 0.2 | 4.63 | 4.01 |
| 6c | 2.9 | 4.60 | 3.98 | 7c | 0.4 | 4.64 | 4.02 |
| 6d | 5.9 | 4.62 | 4.00 | 7d | 0.9 | 4.64 | 4.02 |
| 6e | 8.8 | 4.63 | 4.01 | 7e | 1.9 | 4.64 | 4.02 |
| 6f | 12 | 4.64 | 4.02 | 7f | 7.4 | 4.48 | 3.88 |
| 6g | 12 | 4.71 | 4.08 | 7g | 10 | 4.47 | 3.88 |
| 6h | 18 | 4.62 | 4.00 | 7h | 15 | 4.51 | 3.91 |
| 6i | 23 | 4.63 | 4.01 | ||||
 | ||
| Fig. 3 SAXS pattern of 6h and 7h. | ||
In a detailed investigation of the lattice parameters for the differently dye loaded materials, no significant deviation can be observed. All co-condensed materials possess d100 spacing in a range from 3.88 to 4.03 nm corresponding to lattice parameters ranging from 4.47 to 4.65 nm. In addition, no decrease of the reflection intensity is observed which also suggests that the hexagonal structure is not influenced by the addition of dye. In summary it can be concluded that the applied dye concentrations in a micromolar range do not interfere with the formation of the mesoporous structure as long as the synthesis conditions remain constant.
Transmission electron microscopy (TEM)
In contrast to the gas sorption and SAXS analysis of the hybrid materials where a hexagonal ordering of the silica is deduced from the experimental data, the characterization by transmission electron microscopy (TEM) gives unambiguous evidence for the structural ordering as a direct image of the sample. As shown in Fig. 4, the hexagonal, honeycomb-like ordering of the pores as well as the two-dimensional organization of channels is evident when the sample is viewed in direction of the pores and perpendicular to the pore channels, respectively. The quality of the obtained silica structures are comparable to commercial MCM-41 and show no dependence on the amount of incorporated dye. The assumption of voids inside the hybrid material, as indicated by the hysteresis in the sorption isotherms, can be confirmed in the TEM images. Thus all structural analysis are mutually consistent with each other and confirm the assumed hexagonal mesoporous ordering as well as the absence of an influence of the dye incorporation (in the micromolar range) on the structural ordering. | ||
| Fig. 4 (a) TEM image of 7h along the channel direction and (b) TEM image of 7h perpendicular to the channel direction as well as (c) TEM image of 6i along the channel direction and (d) TEM image of 6i perpendicular to the channel direction. | ||
Excitation and emission properties
 | ||
| Fig. 5 Applied vs. determined concentrations of the hybrid materials 6 and 7 (linear regression analysis gives for grafted: y = 0.585x, R2 = 0.969; and co-condensed: y = 0.371x, R2 = 0.948). | ||
| Grafted | Co-condensed | ||||
|---|---|---|---|---|---|
| Applied conc. of 5 [μmol g−1] | Calculated conc. of 5 [μmol g−1] | Applied conc. of 5 [μmol g−1] | Calculated conc. of 5 [μmol g−1] | ||
| 6a | 1.0 | 0.6 | 7a | 0.1 | 0.04 |
| 6b | 2.5 | 1.5 | 7b | 0.5 | 0.2 |
| 6c | 5.0 | 2.9 | 7c | 1.0 | 0.4 |
| 6d | 10 | 5.9 | 7d | 2.5 | 0.9 |
| 6e | 15 | 8.8 | 7e | 5.0 | 1.9 |
| 6f | 20 | 12 | 7f | 20 | 7.4 |
| 6g | 20 | 12 | 7g | 30 | 11 |
| 6h | 30 | 18 | 7h | 40 | 15 |
| 6i | 40 | 23 | |||
The incorporation of dye into the silica material proceeds more effectively by grafting than by co-condensation. The slopes of the regression lines indicate an incorporation of approximately half of the applied dye concentration in the hybrid material obtained by grafting, whereas for the co-condensed material only one third of the applied concentration is incorporated.
Although a lower concentration of reactive silanol groups can be expected due to calcination of the commercial MCM-41 source a higher degree of incorporation of dye by grafting was proven.
As a consequence the lower applied dye concentration is still sufficient for the grafted materials, in comparison with the equivalent loaded co-condensed hybrids.
Fluorescence quantum yields
The total fluorescence of nine grafted (6) and eight co-condensed (7) hybrid materials in the solid state was determined using an integrating sphere (Ulbricht setup). The quantum yields in dependence on the matrix-embedded dye concentration are shown in Fig. 6 and Table 4. Both types of materials, co-condensed as well as grafted, reveal a similar dependence of the quantum yield on the concentration.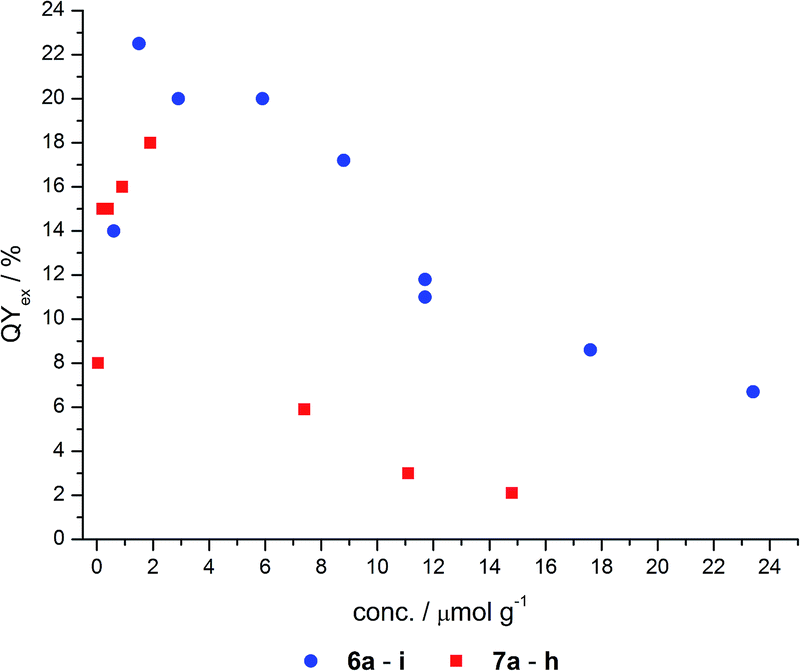 | ||
| Fig. 6 Quantum yields of grafted (6) and co-condensed (7) hybrids. | ||
| Grafted | Co-condensed | ||||
|---|---|---|---|---|---|
| Calculated conc. of 5 [μmol g−1] | Quantum yield | Calculated conc. of 5 [μmol g−1] | Quantum yield | ||
| 6a | 0.6 | 14 | 7a | 0.04 | 8.0 |
| 6b | 1.5 | 23 | 7b | 0.2 | 15 |
| 6c | 2.9 | 20 | 7c | 0.4 | 15 |
| 6d | 5.9 | 20 | 7d | 0.9 | 16 |
| 6e | 8.8 | 17 | 7e | 1.9 | 18 |
| 6f | 12 | 12 | 7f | 7.4 | 5.9 |
| 6g | 12 | 11 | 7g | 11 | 3.0 |
| 6h | 18 | 8.6 | 7h | 15 | 2.1 |
| 6i | 23 | 6.7 | |||
These materials display fluorescence even at very low dye doping levels, such as in the nanomolar range. With increasing dye concentration a steep increase in quantum yield is detected up to a maximum concentration around 2 μmol g−1. Quantum yields of 18 and 23% for the co-condensed and grafted material, respectively, were determined. At higher dye loading the quantum yields decrease even below the quantum yields for the nanomolar dye doping levels. This asymptotic convergence to a completely quenched fluorescence very likely arises from the fluorescence quenching of NR at higher concentrations, as reported for free NR in the solid state.
Although a similar dependence of the quantum yield on the dye loading can be observed, the maximum quantum yield of the co-condensed material 7 is about 4% less than for the grafted material 6. Moreover, the grafted hybrids reach a quantum yield of 7% even at high concentrations, whereas the co-condensed materials are nearly quenched (2%). A possible explanation for this behavior could be the weak acidity of the silanol groups (pKa ≈ 7.1),45 which can protonate the NR molecules, and thereby cause fluorescence quenching.
If the functionalization of the co-condensed hybrids 7 can be assumed to take place preferentially inside the pores, the fluorophore molecules will face an environment with a higher effective concentration of silanol groups in comparison to predominantly outside bound dyes as expected for the grafted case 6. Thus, the dye molecules in co-condensed hybrids 7 will experience a higher degree of protonation and quenching of the NR luminophore compared to the grafted hybrids 6.46
Solvatochromism
The solvatochromic properties of the 2-hydroxy-substituted NR derivative 3 are not significantly affected by functionalization to give the precursor molecule 5. The excitation as well as emission maxima remain almost unchanged, except for the measurement in diethyl ether where the maxima of the precursor molecule 5 are slightly hypsochromically shifted relative to the 2-hydroxy substituted NR 3 (Fig. 7). | ||
| Fig. 7 Excitation and emission maxima of 2-hydroxy substituted NR (3) and precursor molecule 5. | ||
Upon incorporation of the NR derivative 5 into mesoporous silica materials (as studied for hybrid material 7e) the solvatochromic behavior is mostly retained (Fig. 8). However, it was found that the hybrid materials show a red-edge excitation shift (REES) known from polar dyes in viscous solvents.47–50 Thus, since it was not possible to obtain absorption spectra in the solid state and, except for DMSO, in suspension, excitation spectra were recorded for the determination of the absorption maxima. But due to the previous discussion it was not possible to correct these excitation spectra for the inner filter effect. It cannot be ruled out that the measured spectroscopic data are slightly error-prone. However, these systematic errors become negligible as emission spectra were always recorded at excitation in the short wavelength part of the excitation spectra where no shift of the emission spectra could be observed (vide infraFig. 16).
 | ||
| Fig. 8 Solvatochromism of 7e. | ||
Upon changing the environment of the dye the spectroscopic properties are also influenced. The excitation as well as emission maxima of hybrid material 7e are bathochromically shifted in comparison to precursor 5 (Fig. 9 and 10).
 | ||
| Fig. 9 Emission maxima of 5 and 7e in different solvents (grey line at 643 nm represents the emission of the hybrid material in the solid state). | ||
 | ||
| Fig. 10 Excitation maxima of 5 and 7e in various solvents (grey line at 580 nm represents the excitation of the hybrid material in the solid state). | ||
Most strikingly, both spectra reveal a much smaller range of emission and excitation maxima for the hybrid material than for the precursor molecule 5. Whereas the emission of precursor 5 shifts by 4000 cm−1 from hexane to water, the shift decreases to about 2000 cm−1 for the hybrid material 7e. This can be explained by the dominance of the siloxy environment in the pores, which is only affected to a minor extend upon changing the solvent polarity in the pores. This assumption is additionally supported by the emission maxima of the hybrid material in the solid state as indicated by the grey line at 643 nm in Fig. 9. Only the emission of hybrid material 7e suspended in ether or ester causes a hypsochromic shift of the emission maximum and the suspension in water leads to a significant bathochromic shift relative to the emission of 7e in solid state. The redshift for the hybrid material in water relative to the emission in the solid state can be rationalized by extensive hydrogen bonding, leading to a better stabilization of the excited state. The reverse is true for hydrogen-bond accepting solvents, such as ethers and esters which induce a blue shift of the emission.
The effect of the solvent polarity on the excitation spectra is not strongly pronounced, possibly due to the lower sensitivity of the electronic ground state to polarity changes. However, slight deviations due to an inner filter effect have to be taken into account.
Furthermore, the excitation as well as emission spectra of precursor 5 and hybrid material 7e do not change for measurements in methanol, DMSO and water. In all three cases the environment created by the silica matrix is comparable to the environment of the dye in the respective solvent. This is also reflected in the differences in the Stokes shifts of the precursor molecule 5 and the hybrid material 7e (Fig. 11).
 | ||
| Fig. 11 Difference between Stokes shifts of precursor molecule 5 and hybrid material 7e. | ||
Some solvents hardly show any change in the Stokes shift of the hybrid material 7e compared to precursor molecule 5, although their excitation and emission maxima vary significantly. A striking example is the identical Stokes shift of precursor 5 (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1600 cm−1) and hybrid material 7e ( = 1600 cm−1) in hexane, although the emission and excitation of hybrid 7e is shifted bathochromically by 3100 cm−1 in comparison to compound 5 (Fig. 9 and 10). Thus, it can be concluded that the incorporation of the dye into a silica matrix significantly changes the spectroscopic properties but the influence of the solvent polarity on these properties is comparable for both the free precursor 5 and the hybrid material 7e.
= 1600 cm−1) and hybrid material 7e ( = 1600 cm−1) in hexane, although the emission and excitation of hybrid 7e is shifted bathochromically by 3100 cm−1 in comparison to compound 5 (Fig. 9 and 10). Thus, it can be concluded that the incorporation of the dye into a silica matrix significantly changes the spectroscopic properties but the influence of the solvent polarity on these properties is comparable for both the free precursor 5 and the hybrid material 7e.
Comparing the excitation spectra of the differently loaded hybrid materials the effect of loading levels on the excitation maxima for the grafted (6) and co-condensed (7) hybrids can be considered to be only minimal. For instance the excitation maximum of grafted material 6a at 589 nm is shifted bathochromically for material 6c by approximately 100 cm−1 accompanied by a decrease in intensity. The co-condensed hybrids 7 show a similar trend by redshifting from 572 (7c) to 579 nm (7e) (Fig. 12) by approximately 200 cm−1 concomitantly showing a decrease in excitation intensity with increasing concentration. In addition, the shape of the excitation spectra is altered as at the red-edge a relative sharp signal arises whereas the intensity at 590 nm (grafted hybrids 6) and 580 nm (co-condensed hybrids 7) decreases even further. This effect is attributed to the primary inner filter effect leading to an attenuation of the excitation light and resulting in a virtual redshift at high dye concentrations.
 | ||
| Fig. 12 Excitation spectra of selected grafted (6) and co-condensed (7) hybrid materials with variable dye loading (λemission = 700 nm). | ||
The emission spectra indicate a pronounced red shift by 1000 cm−1 upon increase of the dye concentration from 10−7 over 10−6 to 10−5 mol g−1 (Fig. 13). This bathochromic shift is identical for grafted (6) and co-condensed (7) materials as indicated by almost superimposable spectra at comparable dye loading.
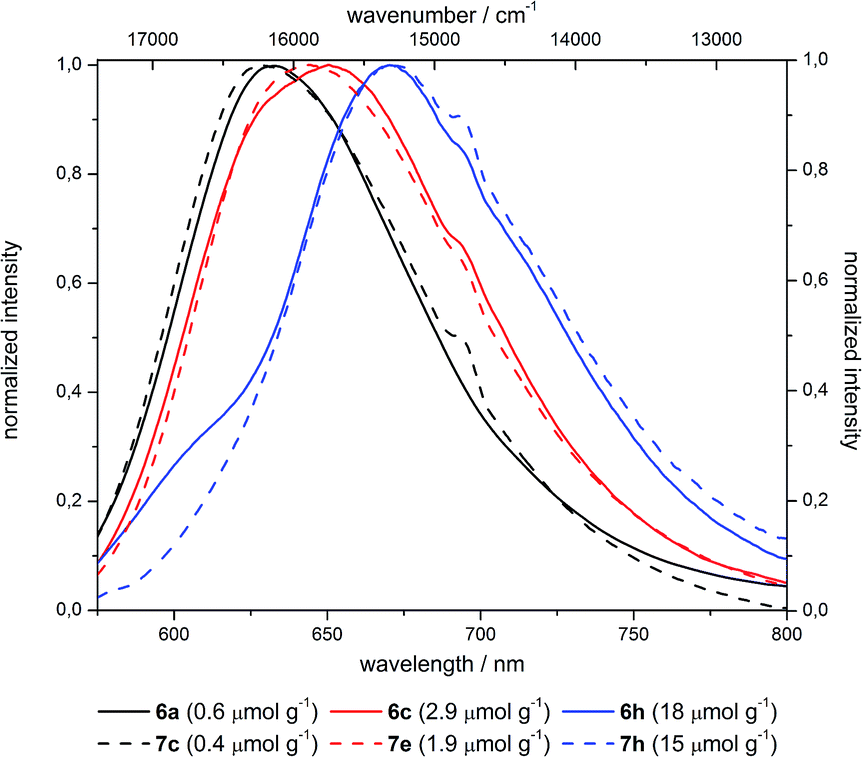 | ||
| Fig. 13 Emission spectra of selected differently loaded grafted (6) and co-condensed (7) hybrid materials (shoulder at 695 nm is a measurement caused artefact). | ||
Thus, this bathochromic shift can be attributed to reabsorption of the hypsochromic part of the emission at higher concentrations by dyes with small Stokes shifts.47 Although NR in general exhibits a large Stokes shift after incorporation in silica such reabsorption events become possible (Fig. 14).51
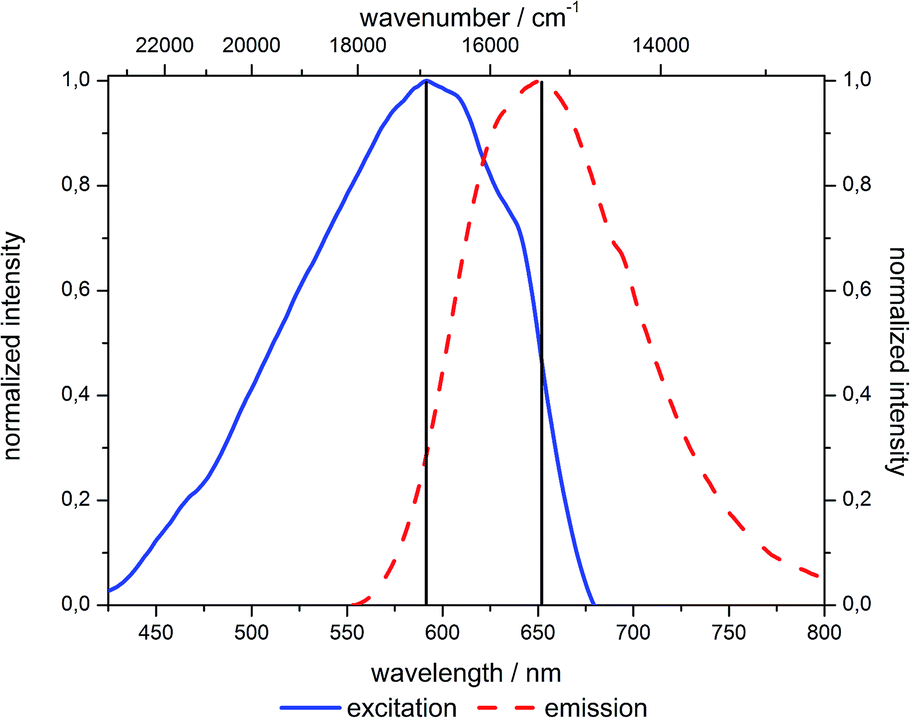 | ||
| Fig. 14 Excitation and emission spectra of 6c (in the solid state). | ||
There is no evidence for the formation of aggregates although the excitation spectra display a different shape and a slight red shift at higher dye concentrations. These findings could imply the forming of aggregates, but considering the spectroscopic data from reports on NR aggregation this scenario seems unlikely in our case.28,51–54 If the alteration of shape in the excitation spectra was caused by aggregation, it would be quite unlikely that for the grafted and co-condensed materials, despite their very different synthesis conditions, nearly identical aggregates would be formed. Furthermore, literature reported aggregates of NR adsorbed in MCM-41 materials and suspended in dichloromethane possess full width at half maximum (fwhm) values of their emission band around 1000 cm−1.54 Even though these values are quite high, they become possible due to higher disorder in a restricted environment leading to a broadening of the emission band.55,56 But comparing the fwhm values of the synthesized materials which are around 1500 cm−1 for hybrids suspended in dichloromethane and even 2000 cm−1 in solid state, aggregation seems even more unlikely. In addition there is no reduction of fwhm at high dye loading levels, relative to the low loaded hybrid material.
Moreover, there is no change in the absorption spectra of the suspensions of hybrid materials in DMSO, except for a shoulder at 640 nm (Fig. S25 and S26, ESI†) for some hybrid materials which can be attributed to protonated NR species. This is additionally supported by fluorescence quenching studies. Therefore, reabsorption effects in the solid state causing a bathochromic shift are by far more plausible than aggregate formation.46,57
The effect of REES can be found for the hybrid materials suspended in different solvents as well as in the solid state, although less pronounced. For hybrid materials 6f and 7g the emission shifts bathochromically by 133 and 88 cm−1, respectively, upon excitation at 546 and 664 nm. This effect is much more pronounced for the hybrid materials in suspension as shown in Fig. 15, with maximum shifts of about 1000 cm−1. It is striking that predominantly the grafted material shows bigger shifts compared to the co-condensed hybrids and that nearly no REES can be observed in water.
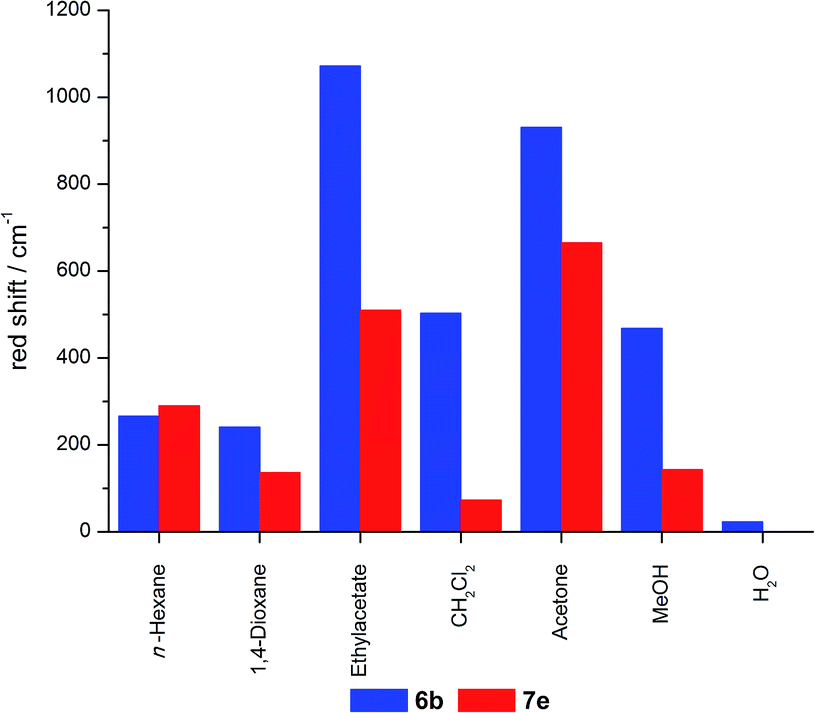 | ||
| Fig. 15 REES of 6b and 7e suspended in different solvents (range of excitation n-hexane: 3333 cm−1, 1,4-dioxane: 2389 cm−1, ethylacetate: 1374 cm−1, CH2Cl2: 3607 cm−1, acetone: 3871 cm−1, MeOH: 4127 cm−1, H2O: 2309 cm−1). | ||
A thorough inspection of the REES of the grafted (6b) and co-condensed (7e) hybrid materials suspended in acetone clearly shows that at excitation wavelengths lower than 540 nm both materials do not distinctly shift the emission in dependence of the excitation wavelength (Fig. 16). Only upon excitation in the red-edge a shift in the emission maxima can be observed.
 | ||
| Fig. 16 REES of 6b and 7e in acetone. | ||
As all grafted materials show a more pronounced REES it can be assumed that the distribution of dye molecules is more inhomogeneous. While some quite unrestricted molecules are located on the outside surface, some relatively restricted fluorophores reside inside the pore channels, and a fraction of molecules which is bound to the pore openings, where high dye concentrations are present, enhancing their restrain. In contrast, the relatively homogenous distribution inside the pores with only slight changes in the restriction of the molecules inside the channels could give rise to a less pronounced REES for the co-condensed materials.
Fluorescence quenching
For further investigation of the different hybrid materials fluorescence quenching experiments were conducted. Upon addition of hydrochloric acid to the water suspended hybrid materials 7e and 6b, a decrease in fluorescence intensity can be monitored (Fig. 17). | ||
| Fig. 17 Fluorescence spectra of water suspended hybrid materials 7e (solid lines) and 6b (dashed lines) upon addition of HCl at the indicated concentrations. | ||
Both materials show an emission maximum at 664 nm with a small shoulder at 720–750 nm in the emissive state. Upon addition of an acid to the samples the emission maximum at 664 nm decreases and slightly shifts bathochromically to 670 nm for high acid concentrations. For the two materials with comparable dye loading levels of 1.5 and 1.9 μmol g−1 the emission spectra at low acid concentrations (up to 6.00 mmol L−1) look very similar. But upon further increase of the acid concentration, the co-condensed material 7e is more efficiently quenched than the grafted material 6b. For the identical relative fluorescence intensity for the co-condensed hybrid 7e a smaller acid concentration is need than for the grafted hybrid 6b (Fig. 18). This indicates the presence of unequally distributed dye loading inside the different materials, which can also be observed in the plot of fluorescence intensities against acid concentrations.
 | ||
| Fig. 18 Stern–Volmer plots of water suspended hybrid materials 6b (blue circles) and 7e (red squares) upon addition of HCl at different concentrations. | ||
In this Stern–Volmer plot the curves deviate from linearity with increasing acid concentrations towards the x-axis which is indicative for the presence of more than one species of fluorophores with variable accessibility to the quencher. These different classes of fluorophores can also be deduced from the excitation spectra of the quenched fluorophores (Fig. 19).
 | ||
| Fig. 19 Excitation spectra of water suspended hybrid materials 6b (dashed lines) and 7e (solid lines) upon addition of HCl at the indicated concentrations (λemission = 720 nm). | ||
In the emissive state the different materials show slightly variable excitation maxima. For the co-condensed material 7e the maximum lies at 602 nm, whereas the maximum of the grafted hybrid 6b is slightly shifted bathochromically to 608 nm. Upon addition of 200 mmol L−1 of acid both materials show the same two excitation maxima at 588 and 640 nm, but in the case of the grafted material 6b the signal at 640 nm is significantly more intense than in the co-condensed case 7e. This newly arising redshifted excitation maximum at 640 nm corresponds to the literature known absorption maximum of NR-H+.46,57
As the grafted materials show a more intense signal of the protonated NR species NR-H+ than in the co-condensed case it can be assumed that the degree of NR-H+ is higher in the grafted hybrid 6b than in the co-condensed 7e. This can be rationalized by preferred functionalization with the dye molecules on the outside surface and the pore openings of grafted materials enabling an easier protonation. In turn, in the co-condensed material the dye molecules are incorporated within the pores or even within the silica walls to a larger extent. In addition the surrounding silica matrix can act as a proton buffer.
Conclusions
The fluorescence dye 2-hydroxy NR was functionalized via propargylation and CuAAC reaction with an azido substituted triethoxysilane derivative to furnish a luminescent precursor for covalent ligation. The formation of mesoporous hybrid materials by incorporation into silica matrices with variable dye loading was accomplished by two synthetic approaches, i.e. by grafting onto a mesoporous MCM-41 host or by in situ co-condensation with concomitant formation of the mesoporous silica-dye hybrid. The structural and optical properties of these novel hybrids were thoroughly studied. Analysis of the materials by nitrogen sorption measurements in combination with SAXS and TEM confirmed a two-dimensional hexagonal columnar ordered mesoporous structure which is not affected by functionalization with the NR dye in the applied micromolar concentrations. Investigation of the optical properties revealed a red-edge excitation shift (REES) and fluorescence quenching, which were utilized to analyze the dye distribution inside the different materials. Therefore it can be qualitatively assumed that different dye species are present inside the grafted and co-condensed materials. While grafted materials are predominantly functionalized on the outside surface and in pore openings at high dye concentrations, the co-condensed materials obviously possess a more homogenous dye distribution and predominant functionalization inside the pores or silica walls. The hybrid materials retain, although much less pronounced, some optical characteristics of the NR, such as the solvatochromism. However, in contrast to the native dye, these synthesized hybrids possess quantum yields of about 20% in the solid state and even fluorescence in aqueous media which opens new alleys to applications of NR-based hybrid materials. Further studies directed to access novel functional organic hybrid materials by this approach are currently underway.Experimental
General considerations
Reagents, catalyst and solvents were purchased reagent grade and used without further purification. Products were purified with column chromatography on silica gel 60 (0.040–0.063 mm) from M&N using flash technique.1H, 13C and 135-DEPT NMR spectra were recorded on a Bruker AVIII-300 and the resonance of the particular solvent was locked as internal standard (CDCl3: 1H δ = 7.26, 13C δ = 77.0, DMSO-d6: 1H δ = 2.50, 13C δ = 39.52). MALDI mass spectra were measured on a Bruker Daltronics Ultraflex I spectrometer, HR-ESI mass spectra were measured on a UHR-QTOF maXis 4G Bruker Daltronics and GC mass spectra were obtained on a GCMS-QP2010 S spectrometer from Shimadzu. Combustion analyses were carried out on Elementar vario MICRO CUBE in the microanalytical laboratory of the Institut für Pharmazeutische und Medizinische Chemie at the Heinrich-Heine-Universität Düsseldorf. IR spectra were recorded on Shimadzu IRAffinity-1. Melting points were measured on a Büchi Melting Point B-540. N2-adsorption–desorption isotherms were determined at 77 K by a Quantachrome Nova 4200e sorption analyser. Prior to the measurements the samples were degassed at 80.0 °C for 20 h. Specific surface areas were calculated using the Brunauer–Emmett–Teller (BET) equation in the low pressure interval p/p0 < 0.3. Pore size distributions were calculated from the adsorption branch using the non-local density function theory based on a slit-pore model. The total pore volume was calculated at the point of p/p0 = 0.95. X-ray diffraction data were collected on a Bruker AXS Nanostar C. The radiation source was a Siemens X-ray canal with a power of 1500 W. The nickel-filtered monochromatic CuKα1 radiation wavelength of 1.5405 nm was maintained by using crossed Göbel mirrors as monochromator. Real-time detection was enabled using a Bruker HI-Star detector. Samples were measured in Hilgenberg glass capillaries with an outer diameter of 0.7 nm. Pre-measurement calibration was carried out at 298 K with silver behenate as a standard reference. Data analysis was carried out using the following software: SAXS from Bruker, Datasqueeze (v. 2.2.8) from Heiney, QTIPlot (v.0.9.8) from Ion Vasilief and LCDiXray (v.1.0) from Golbert. Mesoporous structures were probed by HR-TEM at the institute of microstructure research in Jülich using a FEI TECNAI G2 operated at 200 kV. All HR-TEM samples were prepared by placing a drop of the hybrid material suspension in dichloromethane onto a carbon-coated grid at room temperature.
Excitation and emission spectra were recorded on a Hitachi F-7000 fluorescence spectrophotometer at T = 293 K. Excitation of fluorescence was always carried out at the excitation maximum. Quantum yield determinations of the hybrid powders were obtained with an integrating sphere. Data analysis and quantum yield calculations were performed with the software FL Solutions Version 4.0 by Hitachi.
Synthetic procedures
1H NMR (300 MHz, CDCl3): δ = 0.64–0.71 (m, 2H, –SiCH2–), 1.23 (t, 3JHH = 7.0 Hz, 9H, Si(OCH2CH3)3), 1.65–1.77 (m, 2H, –SiCH2CH2–), 3.26 (t, 3JHH = 7.0 Hz, 2H, –SiCH2CH2CH2–), 3.82 (q, 3JHH = 7.0 Hz, 6H, Si(OCH2CH3)3). 13C NMR (75 MHz, CDCl3): δ = 7.6 (CH2), 18.3 (CH3), 22.7 (CH2), 53.8 (CH2), 58.5 (CH2). GC-MS: m/z = 163 [(C3H9N3O3Si)+], 119 [(C3H7O3Si)+], 79 [(HO)3Si+].
1H NMR (300 MHz, DMSO-d6): δ = 1.27 (t, 3JHH = 7.1 Hz, 6H, –CH3), 3.82 (m, 4H, –CH2–), 6.75 (d, 3JHH = 2.2 Hz, 1H, 6-H), 7.21 (dd, 3JHH = 10.4 Hz, J = 2.2 Hz, 1H, 4-H), 7.55 (d, 3JHH = 10.4 Hz, 1H, 3-H).
:1) to give 9-(diethylamino)-2-hydroxy-5H-benz[a]phenoxazin-5-one (3) (8.66 g, 66%) as a violet solid, Mp 298 °C.
1H NMR (300 MHz, DMSO-d6): δ = 1.14 (t, 3JHH = 7.0 Hz, 6H, –CH3), 3.45 (q, 3JHH = 7.1 Hz, 4H, –CH2–), 6.11 (s, 1H, 6-H), 6.57 (d, 4JHH = 2.6 Hz, 1H, 8-H), 6.74 (dd, 3JHH = 9.1 Hz, 4JHH = 2.7 Hz, 1H, 10-H), 7.07 (dd, 3JHH = 8.6 Hz, 4JHH = 2.5 Hz, 1H, 3-H), 7.52 (d, 3JHH = 9.1 Hz, 1H, 11-H), 7.85 (d, 4JHH = 2.4 Hz, 1H, 1-H), 7.95 (d, 3JHH = 8.6 Hz, 1H, 4-H), 10.41 (s, 1H, –OH). 13C NMR (75 MHz, DMSO-d6): δ = 12.4 (CH3), 44.4 (CH2), 96.0 (CH), 104.0 (CH), 108.1 (CH), 109.8 (CH), 118.3 (CH), 123.8 (Cquat), 127.4 (CH), 130.7 (CH), 133.7 (Cquat), 138.6 (Cquat), 146.3 (Cquat), 150.6 (Cquat), 151.5 (Cquat), 160.5 (Cquat), 181.5 (Cquat). IR: /cm−1 = 638 (m), 671 (m), 687 (m), 702 (m), 745 (m), 783 (m), 797 (m), 818 (m), 847 (m), 887 (m), 910 (m), 953 (m), 1013 (m), 1028 (m), 1028 (m), 1045 (m), 1074 (m), 1112 (vs), 1150 (m), 1159 (m), 1180 (m), 1221 (m), 1258 (vs), 1288 (m), 1317 (vs), 1377 (m), 1406 (m), 1439 (m), 1474 (m), 1485 (m), 1505 (m), 1520 (m), 1537 (m), 1562 (m), 2625 (br), 2870 (w), 2924 (w), 2963 (w), 3046 (br). HR-MS (ESI) calcd for (C20H18N2O3 + H)+: m/z = 335.13902 (100%), 336.14237 (22%); found: 335.13893 (100%), 336.14204 (15%). Anal. calcd for C20H18N2O3 (334.4): C 71.84, H 5.43, N 8.38; found: C 71.66, H 5.52, N 8.09.
:1, with 5% triethylamine) to give compound 4 as a red solid (89.0 mg, 24%), Mp 192–194 °C.
1H NMR (300 MHz, CDCl3): δ = 1.25 (t, 3JHH = 7.1 Hz, 6H, –NCH2CH3), 2.58 (t, 4JHH = 2.4 Hz, 2H, –OCH2CCH), 3.45 (q, 3JHH = 7.1 Hz, 4H, –NCH2CH3), 4.88 (t, 4JHH = 2.4 Hz, 1H, –OCH2–), 6.28 (s, 1H, 6-H), 6.42 (d, 4JHH = 2.6 Hz, 1H, 8-H), 6.62 (dd, 3JHH = 9.1 Hz, 4JHH = 2.6 Hz, 1H, 10-H), 7.22 (dd, 3JHH = 8.7 Hz, 4JHH = 2.6 Hz, 1H, 3-H), 7.57 (d, 3JHH = 9.1 Hz, 1H, 11-H), 8.10 (d, 4JHH = 2.6 Hz, 1H, 1-H), 8.23 (d, 3JHH = 8.7 Hz, 1H, 4-H). 13C NMR (75 MHz, CDCl3): δ = 12.6 (CH3), 45.1 (CH2), 56.1 (CH2), 96.3 (CH), 105.3 (CH), 107.2 (CH), 109.6 (CH), 118.4 (CH), 124.8 (Cquat), 126.4 (Cquat), 127.9 (CH), 131.2 (CH), 134.0 (Cquat), 139.8 (Cquat), 146.8 (Cquat), 150.8 (Cquat), 152.1 (Cquat), 160.1 (Cquat), 183.1 (Cquat). IR: /cm−1 = 679 (m), 591 (m), 704 (m), 743 (m), 756 (m), 789 (m), 807 (m), 820 (s), 845 (m), 864 (m), 880 (m), 893 (m), 924 (m), 1005 (s), 1034 (m), 1080 (s), 1115 (s), 1150 (m), 1179 (m), 1194 (m), 1207 (m), 1250 (s), 1267 (s), 1292 (m), 1315 (m), 1335 (m), 1373 (m), 1404 (s), 1454 (m), 1468 (m), 1491 (m), 1518 (w), 1530 (w), 1555 (m), 1572 (m), 1589 (s), 1638 (w), 1687 (w), 1726 (m), 2106 (w), 2332 (w), 2359 (w), 2872 (w), 2928 (m), 2959 (m), 3194 (m). MALDI-TOF: m/z = 373.4 (C23H20N2O3 + H)+.
:1) to give compound 5 as a red solid (60.0 mg, 45%).
1H NMR (300 MHz, CDCl3): δ = 0.61 (m, 2H, –NCH2CH2CH2Si–), 1.24 (m, 15H, Si(OCH2CH3)3, –NCH2CH3), 2.05 (m, 2H, –NCH2CH2CH2Si–), 3.47 (q, 3JHH = 7.1 Hz, 4H, –NCH2CH3), 3.80 (q, 6H, Si(OCH2CH3)3), 4.39 (t, 3JHH = 7.2 Hz, 2H, –NCH2CH2CH2Si–), 5.42 (s, 2H, –OCH2C–), 6.30 (s, 1H, 6-H), 6.45 (d, 4JHH = 2.6 Hz, 1H, 8-H), 6.66 (dd, 3JHH = 9.1 Hz, 4JHH = 2.7 Hz, 1H, 10-H), 7.23 (dd, 3JHH = 8.7 Hz, 4JHH = 2.5 Hz, 1H, 3-H), 7.60 (d, 3JHH = 9.1 Hz, 1H, 11-H), 7.69 (s, 1H, –CCHN–), 8.18 (d, 4JHH = 2.5 Hz, 1H, 1-H), 8.23 (d, 3JHH = 8.7 Hz, 1H, 4-H). 13C NMR (75 MHz, CDCl3): δ = 7.47 (CH2), 12.6 (CH3), 18.3 (CH3), 24.2 (CH2), 45.1 (CH2), 52.6 (CH2), 58.5 (CH2), 62.5 (CH2), 96.3 (CH), 105.3 (CH), 106.9 (CH), 109.6 (CH), 118.4 (CH), 122.8 (CH), 124.8 (Cquat), 126.1 (Cquat), 127.9 (CH), 131.2 (CH), 134.1 (Cquat), 139.8 (Cquat), 143.5 (Cquat), 146.9 (Cquat), 150.8 (Cquat), 152.1 (Cquat), 160.9 (Cquat), 183.2 (Cquat). IR: /cm−1 = 638 (m), 669 (m), 683 (m), 716 (m), 789 (s), 818 (s), 881 (m), 910 (m), 953 (m), 1011 (m), 1030 (m), 1072 (vs), 1111 (vs), 1161 (m), 1182 (m), 1194 (m), 1223 (m), 1256 (s), 1271 (s), 1314 (s), 1346 (m), 1406 (s), 1439 (m), 1458 (m), 1470 (m), 1479 (m), 1497 (m), 1520 (m), 1558 (m), 1582 (vs), 1620 (m), 1641 (m), 1670 (w), 1680 (w), 2891 (w), 2928 (w), 2972 (w), 3084 (w), 3134 (w). HR-MS (ESI) calcd for (C32H41N5O6Si + H)+: m/z = 620.28989; found: 620.28975; calcd for (C26H29N5O6Si + H)+: m/z = 536.19599; found: 536.19595.
Synthesis of hybrid materials
| Sample | Applied loading of hybrid with 5 [μmol g−1] | Determined loading of hybrid with 5 [μmol g−1] | Volume of stock solution [mL] | Mass of MCM-41 [g] | Volume of ethanol [mL] | Yield of NR grafted MCM-41 hybrid materials 6 [g] |
|---|---|---|---|---|---|---|
| 6a | 1.0 | 0.6 | 8.91 of c1(5) | 0.437 | 6.10 | 0.408 |
| 6b | 2.5 | 1.5 | 0.50 of c3(5) | 1.00 | 14.5 | 0.560 |
| 6c | 5.0 | 2.9 | 14.3 of c2(5) | 1.75 | 0.70 | 1.64 |
| 6d | 10 | 5.9 | 2.00 of c3(5) | 1.00 | 13.0 | 0.624 |
| 6e | 15 | 8.8 | 3.00 of c3(5) | 1.00 | 12.0 | 0.752 |
| 6f | 20 | 12 | 3.50 of c4(5) | 1.00 | 11.5 | 0.802 |
| 6g | 20 | 12 | 14.3 of c2(5) | 0.437 | 0.70 | 0.396 |
| 6h | 30 | 18 | 5.24 of c4(5) | 1.00 | 9.76 | 0.804 |
| 6i | 40 | 23 | 6.99 of c4(5) | 1.00 | 8.01 | 0.892 |
:x:0.140:2.40:2.00:100 (molar ratios x of precursor 5 are given in Table 6). First the template C16TMABr was dissolved in deionized water and ethylamine, which was employed as a 70 wt% aqueous solution before TEOS and the molar amount x of precursor 5 in methanol were added. For the syntheses of materials 7f–h 10 molar equivalents of methanol were used. The mixtures were stirred at room temperature for 24 h with a speed of 750 rpm before they were heated to 100 °C for 24 h. The obtained suspensions were centrifuged (10 min, 4000 rpm), decanted and washed with ethanol before they were centrifuged again. Then the residues were suspended in ethanol (80 mL) and concentrated aqueous hydrochloric acid solution (2 mL) and stirred at 80 °C for 24 h. The mixtures were centrifuged, and the solids were transferred into a Soxhlet extraction thimble and extracted with ethanol over a period of 48 h. The obtained powders were washed once with triethylamine (2 mL) and ethanol (80 mL) upon which they turned red again. The solids were washed with ethanol (3 × 80 mL) and centrifuged as described above till the supernatant reached pH 7. The obtained violet powders were dried at 60 °C and 10−3 mbar for 3 days to mass constancy.
| Sample | Applied loading of hybrid with 5 [μmol g−1] | Determined loading of hybrid with 5 [μmol g−1] | Molar ratio x of precursor 5 | Yield of co-condensed NR hybrid materials 7 [g] |
|---|---|---|---|---|
| 7a | 0.1 | 0.04 | 2.08 × 10−5 | 2.23 |
| 7b | 0.5 | 0.2 | 1.04 × 10−4 | 2.01 |
| 7c | 1.0 | 0.4 | 2.08 × 10−4 | 2.86 |
| 7d | 2.5 | 0.9 | 5.20 × 10−4 | 2.83 |
| 7e | 5.0 | 1.9 | 1.04 × 10−3 | 2.87 |
| 7f | 20 | 7.4 | 4.16 × 10−3 | 1.23 |
| 7g | 40 | 11 | 6.24 × 10−3 | 1.47 |
| 7h | 30 | 15 | 8.32 × 10−3 | 1.41 |
Acknowledgements
The authors cordially thank the Strategic Research Fund of the Heinrich Heine University, the Deutsche Forschungsgemeinschaft DFG (Mu 1088/9-1), and the Fonds der Chemischen Industrie for financial support. Furthermore we thank Dr Juri Barthel and the Ernst Ruska-Centre (ER-C) for help and access to the HR-TEM facilities.Notes and references
- C. T. Kresge, M. E. Lenowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- C. T. Kresge, J. C. Vartuli, W. J. Roth and M. E. Leonowicz, Stud. Surf. Sci. Catal., 2004, 148, 53 CrossRef CAS.
- C. T. Kresge and W. J. Roth, Chem. Soc. Rev., 2013, 42, 3663 RSC.
- A. Taguchi and F. Schüth, Microporous Mesoporous Mater., 2005, 77, 1 CrossRef CAS.
- A. B. Descalzo, M. D. Marcos, C. Monte, R. Martínez-Máñez and K. Rurack, J. Mater. Chem., 2007, 17, 4716 RSC.
- G. Wirnsberger, B. J. Scott and G. D. Stucky, Chem. Commun., 2001, 119 RSC.
- F. Marlow, M. D. McGehee, D. Zhao, B. F. Chmelka and G. D. Stucky, Adv. Mater., 1999, 11, 632 CrossRef CAS.
- N. Vadia and S. Rajput, Asian J. Pharm. Clin. Res., 2011, 4, 44 CAS.
- V. Mameava, C. Sahlgren and M. Lindén, Adv. Drug Delivery Rev., 2013, 65, 689 CrossRef PubMed.
- J. M. Rosenholm, C. Sahlgren and M. Lindén, Nanoscale, 2010, 2, 1870 RSC.
- Y.-S. Lin, C.-P. Tsai, H.-Y. Huang, C.-T. Kuo, Y. Hung, D.-M. Huang, Y.-C. Chen and C.-Y. Mou, Chem. Mater., 2005, 17, 4570 CrossRef CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216 CrossRef CAS PubMed.
- M. Sharifi, R. Marschall, M. Wilhelm, D. Wallacher and M. Wark, Langmuir, 2011, 27, 5516 CrossRef CAS PubMed.
- N. Gartmann and D. Brühwiler, Angew. Chem., Int. Ed., 2009, 45, 6354 CrossRef PubMed.
- D. Aiello, R. Aiello, F. Testa, T. Martino, I. Aiello, M. La Deda and M. Ghedini, J. Photochem. Photobiol., A, 2009, 201, 81 CrossRef CAS.
- C. M. Carbonaro, R. Corpino, P. C. Ricci, D. Chiriu and C. Cannas, AIP Conf. Proc., 2014, 1624, 23 CrossRef CAS.
- G. Schulz-Ekloff, D. Wöhrle, B. van Duffel and R. A. Schoonheydt, Microporous Mesoporous Mater., 2002, 51, 91 CrossRef CAS.
- D. Aiello, A. M. Talarico, F. Teocoli, E. I. Szerb, I. Aiello, F. Testa and M. Ghedini, New J. Chem., 2011, 35, 141 RSC.
- Q. He, J. Shi, X. Cui, J. Zhao, Y. Chen and J. Zhou, J. Mater. Chem., 2009, 19, 3395 RSC.
- P. Greenspan, E. P. Mayer and S. D. Fowler, J. Cell Biol., 1985, 100, 965 CrossRef CAS PubMed.
- N. N. Alekseev, SU 1 109 393, 1983.
- H. Tajalli, A. G. Gilani, M. S. Zakerhamidi and P. Tajalli, Dyes Pigm., 2008, 78, 15 CrossRef CAS.
- J. L. Meinershagen and T. Bein, J. Am. Chem. Soc., 1999, 121, 448 CrossRef CAS.
- G. Hungerford and J. A. Ferreira, J. Lumin., 2001, 93, 155 CrossRef CAS.
- J. Jose and K. Burgess, J. Org. Chem., 2006, 71, 7835 CrossRef CAS PubMed.
- J. Jose, A. Loudet, Y. Ueno, R. Barhoumi, R. C. Burghardt and K. Burgess, Org. Biomol. Chem., 2010, 8, 2052 CAS.
- T. Behnke, C. Würth, K. Hoffmann, M. Hübner, U. Panne and U. Resch-Genger, J. Fluoresc., 2011, 21, 937 CrossRef CAS PubMed.
- T. Felbeck, T. Behnke, K. Hoffmann, M. Grabolle, M. M. Lezhina, U. H. Kynast and U. Resch-Genger, Langmuir, 2013, 29, 11489 CrossRef CAS PubMed.
- J. Qian, X. Li, M. Wie, X. Gao, Z. Xu and S. He, Opt. Express, 2008, 16, 19568 CrossRef CAS PubMed.
- R. Sastre and A. Costela, Adv. Mater., 1995, 7, 198 CrossRef CAS.
- S.-Y. Park, Y. Kubota, K. Funabiki, M. Shiro and M. Matsui, Tetrahedron Lett., 2009, 50, 1131 CrossRef CAS.
- S. A. Martin-Brown, Y. Fu, G. Saroja, M. M. Collinson and D. A. Higgins, Anal. Chem., 2005, 77, 486 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed.
- M. Berchel, J.-P. Haelters, D. Afonso, A. Maroto, L. Deschaps, P. Giamarchi and P.-A. Jaffrès, Eur. J. Org. Chem., 2014, 1076 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169 CrossRef CAS.
- H.-P. Lin, S.-T. Wong, C.-Y. Mou and C.-Y. Tang, J. Phys. Chem. B, 2000, 104, 8967 CrossRef CAS.
- D. Zhao, Y. Wan and W. Zhou, Ordered Mesoporous Materials, Wiley-VCH GmbH & Co. KGaA, Weinheim, 2013 Search PubMed.
- G. A. Tompsett, L. Krogh, D. W. Griffin and W. C. Conner, Langmuir, 2005, 21, 8214 CrossRef CAS PubMed.
- P. Cool, E. F. Vasant and O. Collart, in Inorganic Chemistry in Focus II, ed. G. Meyer, D. Naumann and L. Wesemann, Wiley, Weinheim, 2005, p. 319 Search PubMed.
- M. T. Anderson, J. E. Martin, J. G. Odinek and P. P. Newcomer, Chem. Mater., 1998, 10, 1490 CrossRef CAS.
- S. Liu, P. Cool, O. Collart, P. van der Voort, E. F. Vasant, O. I. Lebedev, G. van Tendeloo and M. Jiang, J. Phys. Chem. B, 2003, 107, 10405 CrossRef CAS.
- Z. Zhou, A. W. Franz, S. Bay, B. Sarkar, A. Seifert, P. Yang, A. Wagener, S. Ernst, M. Pagels, T. J. J. Müller and W. R. Thiel, Chem.–Asian J., 2010, 5, 2001 CrossRef CAS PubMed.
- K. J. Edler, in Porous Materials, ed. D. W. Bruce, D. O'Hare and R. I. Walton, John Wiley & Sons, Ltd, Chinchester, 2011, p. 108 Search PubMed.
- M. L. Hair and W. Hertl, J. Phys. Chem., 1970, 74, 91 CrossRef CAS.
- E. M. Moreno and D. Levy, Chem. Mater., 2000, 12, 2334 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Baltimore, 3rd edn, 2006 Search PubMed.
- A. P. Demchenko, Luminescence, 2002, 17, 19 CrossRef CAS PubMed.
- S. Mukherjee, H. Raghuraman and A. Chattopadhyay, Biochim. Biophys. Acta, 2007, 59 CrossRef CAS PubMed.
- R. Saxena, S. Shrivastava, S. Haldar, A. S. Klymchenko and A. Chattopadhyay, Chem. Phys. Lipids, 2014, 183, 1 CrossRef CAS PubMed.
- E. Fleige, B. Ziem, M. Grabolle, R. Haag and U. Resch-Genger, Macromolecules, 2012, 45, 9452 CrossRef CAS.
- T. Behnke, C. Würth, E.-M. Laux, K. Hoffmann and U. Resch-Genger, Dyes Pigm., 2012, 94, 247 CrossRef CAS.
- R. Varghese and H.-A. Wagenknecht, Chem.–Eur. J., 2010, 16, 9040 CrossRef CAS PubMed.
- C. Martín, P. Piatowski, B. Cohen, M. Cil, M. T. Navarro, A. Corma and A. Douhal, J. Phys. Chem. C, 2015, 119, 13283 Search PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- N. L. Selivanov, L. G. Samsonova, V. Y. Artyukhov and T. N. Kopylova, Russ. Phys. J., 2011, 54, 601 CrossRef CAS.
- G. Dördelmann, H. Pfeiffer, A. Birkner and U. Schatzschneider, Inorg. Chem., 2011, 50, 4362 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structural characterization of hybrid materials 6, and 7 (nitrogen sorption, SAXS, and TEM), spectroscopic characterization of precursor 5 and hybrid materials 6 and 7 (UV/Vis and fluorescence), red-edge excitation shift of hybrid materials 6b and 7e, fluorescence quenching of hybrid materials 6b and 7e. See DOI: 10.1039/c5ra22736d |
| This journal is © The Royal Society of Chemistry 2016 |