
Self-assembled lanthanum hydroxide microspheres within a reaction–diffusion framework: synthesis, characterization, control and application†
Ghida A. Al Akhrass,
Manal Ammar,
Houssam El-Rassy and
Mazen Al-Ghoul*
Department of Chemistry, American University of Beirut, P.O.Box 11-0236, Riad El-Solh 1107 2020, Beirut, Lebanon. E-mail: mazen.ghoul@aub.edu.lb
First published on 23rd December 2015
Abstract
We present the synthesis of spherical microparticles by self-assembly of lanthanum hydroxide nanoplatelets in a reaction–diffusion framework, which consists of diffusing an outer ammonia solution into a hydrogel matrix containing lanthanum ions (inner electrolyte). The coupling of reaction–diffusion with nucleation and growth of crystals leads to the formation of precipitation bands well separated in space. The advantage of this method lies in its simplicity to prepare crystalline lanthanum hydroxide at ambient conditions with average particle sizes with small dispersion and varying between ∼300 nm and ∼70 μm, depending on the band location in space. The control over the size of the particles is furthermore studied as a function of the concentration of the inner electrolyte, the thickness of the gel matrix and temperature. The morphology of the spheres is elucidated and is shown to exhibit fascinating topology as a result of the tight packing of the nanoplatelets. Consequently, we demonstrate the remarkable potential of these spheres to swiftly adsorb Congo red (an azo dye) in aqueous solution.
A. Introduction
The lanthanide series has gained a considerable deal of attention due to the unique 4f electron configuration. Due to their attractive optical, electrical, and magnetic properties, lanthanides have found numerous applications as catalysts,1,2 semiconductors,3 sensors,4 high-quality phosphors,5,6 and time-resolved fluorescence (TRF) biological labels.7,8 More recently, rare earth-doped optoelectronic materials9,10 have been used in the design of high-performance luminescent devices,11,12 made possible because of the fascinating up-conversion properties of lanthanides.13,14 Lanthanum, being the lightest representative of the lanthanides series, has been widely studied in its hydroxide, oxide, oxalate, phosphate, and oxychloride forms. In addition to the applications shared by the lanthanides, lanthanum has been particularly invested in the development of electrode materials,15 solid electrolytes,16 dielectric materials,17 ceramics,18,19 and fuel cell catalysts.20Of the lanthanum forms, lanthanum hydroxide has been heavily used as ceramic, superconductive, catalytic, hydrogen storage, and electrode materials.21 Moreover, the synthesis of lanthanum hydroxide often provides a facile route to the oxide form through a simple thermal dehydration.22,23 The extremely hydrophilic OH groups on their outer surface can serve as active sites for introducing novel functional groups through simple condensation reactions.24 Another useful surface property of lanthanum hydroxide is its huge sorbent capacity for several organic dyes25 and arsenic compounds,26 suggesting a promising application in waste-water treatment.
Various routes have been reported in literature for the synthesis of lanthanum hydroxide, such as the hydrothermal method,27,28 solvothermal approach,29 sonochemical process,30 electrochemical synthesis,31 and co-precipitation procedure.32,33 Other synthetic routes include the non-aqueous sol–gel method,34 electrodeposition,35 and composite-hydroxide-mediated (CHM) approach.36 Most of these synthetic routes have yielded similar morphologies that include nanobelts,36 nanorods,37 nanotubes,38 and nanowires.39 Various attempts have been carried out as well to control the size of lanthanum hydroxide particles to reach the nanometric scale. Such trials are often carried out to take advantage of the novel physical and chemical properties exhibited by nanomaterials that are often unobserved in their bulk counterparts. The control of morphology and size of lanthanum hydroxide has been achieved successfully in many studies by adjusting experimental parameters, such as reagent concentration, reaction time,28 solution pH,27 surfactant addition,30 temperature, and solvent type.40
B. Reaction–diffusion framework
In this work, we present the synthesis of lanthanum hydroxide in a precipitate system designed by using reaction–diffusion of electrolytes within an organic gel template (agar in this work) to avoid sedimentation and hydrodynamic advection. The system is prepared by diffusing the initially segregated co-precipitating components: the outer electrolyte (hydroxide ions from ammonia) is allowed to diffuse into the gel matrix that contains the inner electrolyte (lanthanum ions). As they diffuse, they precipitate by forming numerous solid-containing lanthanum hydroxide bands, separated by clear regions, due to the well-known Liesegang instability.41 We take advantage of this banding phenomenon to prepare and control the size of the solid particles as will be demonstrated later. This method is what we call the reaction–diffusion framework.42 Our novel method of synthesis is advantageous in that it can be easily carried out under facile conditions with a low-cost during relatively short time. Because the reacting components are initially separated and poured one on top of the other, we establish a gradient of supersaturation starting at the gel–solution interface and extending down the tube. Since nucleation and growth (and also ripening) of the solid are highly dependent on supersaturation, nucleation dominates near the interface and we obtain many smaller particles, whereas down the tube where growth dominates we end up with larger particles.43 This gradient of supersaturation thus results in a gradient of particle sizes that are distributed among the bands along the tube. Therefore, we can collect various sizes at different bands or heights along the tube. The bands also follow a spacing law whereby the spacing between consecutive bands increases as we move down the tube. It is also worth noting that the banding might exhibit further secondary structures whereby each band is composed of thinner secondary bands (Fig. 1). They result from nonlinear interactions between the diffusion process and the chemical kinetics of the precipitation process. | ||
| Fig. 1 Band formation for different inner concentrations of lanthanum chloride (A) 50 mM; (B) 200 mM; (C) 500 mM; in 1% agar with fixed outer concentration of 14.8 M ammonia. | ||
Using this framework, we also report the control of the morphology and the size of the particles of lanthanum hydroxide by adjusting the thickness of the gel, the concentrations of the electrolytes and the temperature. Size-selection is also made possible through the obtained space-dependent particle size evolution.
C. Experimental section
1. Preparation of gel and synthesis
All chemical reagents are of analytical grade and used directly without any further purification. Lanthanum(III) chloride heptahydrate (LaCl3·7H2O) is purchased from AnalaR; agar is provided by Bacto; and ammonia is supplied by Fisher Scientific.We prepared a stock solution of lanthanum chloride (1 M). We then weigh the exact mass of agar to obtain the desired gel percentage. We transferred the obtained solution to (200 × 20 mm) tubes in such a way that only two-thirds of the tube was filled. The tubes were then stored in a thermostat at 20 °C to allow the polymerization of agar. After the solidification of agar, which took around 2 hours under the experimental conditions, the outer electrolyte (ammonia) was added above the gel without disturbing it. Immediate precipitation of lanthanum hydroxide took place at the interface.
2. Characterization
To extract the solid, the gel is heated in double distilled water until the entire agar dissolves. The lanthanum hydroxide particles are then separated from solution by centrifugation. Finally, we freeze dry the samples for 12 hours, after which the solid is subject to characterization techniques.Powder XRD spectrum is recorded by a Bruker d8 discover X-ray diffractometer with a Cu-Kα radiation (λ = 1.5405 Å). Scanning electron microscopy (SEM) is used to image platinum-coated samples on a carbon tape using a FEI Quanta 600 FEGSEM instrument. FTIR measurements are carried out on KBr pellets using Thermo Nicolet 4700 Fourier Transform Infrared Spectrometer equipped with a Class 1 laser.
3. Congo red adsorption
Congo red (CR; also known by C.I. 22120) is a diazo dye widely used in textile industry having a formula C32H22N6Na2O6S2 and a molecular weight of 696.66 g mol−1. The removal of CR from simulated wastewater by adsorption onto the microspheres of lanthanum hydroxide under various conditions reveals the potential application of these materials for environmental purposes. In typical experiments, the initial amount of the adsorbent (normally 10.0 mg) was dropped in a glass vial containing the CR solution (normally 20 mL) at set pH under shaking in a Julabo SW 23 controlled-temperature water bath operating at 200 rpm. Aliquots were systematically taken at pre-defined time intervals, centrifuged using a Thermo Scientific Heraeus Pico 17 centrifuge, and the CR concentration was determined by measuring the absorbance of the supernatant solution at the maximum absorption wavelength (λ = 498 nm) using a Thermo Scientific Evolution 300 UV/VIS/NIR spectrophotometer.D. Results and discussion
1. FTIR spectroscopy
In order to assess the vibrational modes of the present functional groups, the collected solid has been studied by Fourier transform infrared (FTIR) spectroscopy (Fig. 2A). The spectrum shows at 3427 cm−1 a characteristic peak of the stretching mode of hydroxyl groups and a distinct sharp peak at 3607 cm−1 that is assigned to the stretching mode of hydroxyl group of free La–OH groups. The band centered at 1634 cm−1 corresponds to the deformation vibration of water molecules. Regarding the three bands shown at 1037 cm−1, 1366 cm−1, and 1502 cm−1, they can be attributed to carbonate ions resulting from the water dissolution of carbon dioxide present in air at high pH. The band at low wavenumber (648 cm−1) can be assigned to the bending mode of the La–O bond of lanthanum hydroxide crystals. Consequently, the FTIR spectrum indicates that the extracted solid from the reaction zone is lanthanum hydroxide. | ||
| Fig. 2 (A) FTIR spectrum of the extracted solids. (B) Powder X-ray diffraction spectrum of the extracted solids. | ||
2. Powder X-ray diffraction
X-ray diffraction measurement is also performed to study the phase purity of the sample. The main peaks along with the corresponding lattice planes assignments are shown in (Fig. 2B). The observed diffraction pattern perfectly matches the hexagonal phase of lanthanum hydroxide, indicating that the synthetic method employed results in highly crystalline material, with negligible crystalline by-products. We can affirm the synthesis of lanthanum hydroxide with hexagonal symmetry, P63/m space group, and cell parameters a = 6.528 Å and c = 3.858 Å (JCPDS no. 36-1481).3. Effect of concentration of inner electrolyte
Different experiments are carried out in which the concentration of the inner electrolyte is varied (50 mM, 200 mM, 500 mM) while those of outer electrolyte and agar gel are kept fixed at 14.8 M and 1% (Fig. 1). To enhance the comparison, each tube with distinct inner concentration is divided into 3 parts (Fig. 3A), from which the solid is extracted and subject for study under SEM. Region (1) is 1 cm from the interface; region (2) is between 1–7.5 cm; and region (3) is between 7.5–14 cm. As can be seen in Fig. 3C, there is a strong concentration dependence of the size and morphology of the synthesized lanthanum hydroxide particles. The average particle size is reported as a mean of a sample size that ranges between 10 and 50 particles using the MIRA TESCAN image analysis software (ESI S2 and S3†). At low concentration of inner electrolyte (LaCl3), spheres of 320 nm in average size are formed near the interface separating the two electrolytes. However, when higher inner concentrations (200 mM, 500 mM) are used, rod-shaped particles are obtained with a length of 1.0 μm and a width of 0.4 μm for a concentration of 200 mM and with a length of 2.5 μm and a width of 0.8 μm for a 500 mM inner concentration (See ESI S1†). In the middle portion of the tube, only spherical particles such as those shown in Fig. 3B are obtained that increase consistently in size from around 2 μm (50 mM) to 9 μm (200 mM) and 63 μm (500 mM). A similar trend is observed near the end of the tube where the spheres increase in size from around 3 μm (50 mM) to 23 μm (200 mM) and 70 μm (500 mM) (Fig. 3C). The data indicate that the size of the spheres increase in parallel with the increase in the concentration of the inner electrolyte. This fact stems from the main characteristic of RDF, where the supersaturation gradient plays the major role in the spatial variation of the particle sizes. Increasing the inner concentration of the electrolyte (La3+) at a fixed outer concentration (NH3) will furnish larger initial supersaturation gradients than those of relatively lower inner concentrations, but with the same trend of decreasing magnitude as we move away from the interface. The increase in magnitude of supersaturation as well as its spatial variation are nonlinear and depend on the kinetics of the precipitation as well as the diffusion coefficients of the reacting species. This clearly explains the conspicuous trend in (Fig. 3C). | ||
| Fig. 3 (A) The tube in which reaction–diffusion takes place is divided in 3 regions: (1) 1 cm from the interface; (2) 1–7.5 cm; (3) 7.5–14 cm. (B) SEM image of the spherically-shaped solid particles extracted from the tube. (C) Effect of the inner concentration on the average diameter size of the spherical particles at fixed outer ammonia concentration [NH3] = 14 M. (D) Effect of the gel concentration on the average particle size for [La3+] = 50 mM and [NH3] = 14 M. (E) Effect of temperature on the average particle size for [La3+] = 50 mM and [NH3] = 14 M. | ||
4. Effect of gel thickness
Another attempt to control the size of the spherical morphology has been made by varying the thickness of the agar matrix (0.5, 1, 1.5, 2%) while keeping the concentrations of inner and outer electrolytes constant at 50 mM and 14.8 M respectively (Fig. 3D). Each tube is also divided to 3 parts to allow for a more precise description. Near the interface, higher gel concentrations produce smaller particle sizes: (1) 0.5%, 570 nm; (2) 1%, 365 nm; (3) 1.5%, 340 nm; (4) 2%, 300 nm. A similar trend is observed in the middle portion of the tube: (1) 0.5%, 3 μm; (2) 1%, 2 μm; (3) 1.5%, 1.5 μm; (4) 2%, 1 μm. A strong gel-dependence of size is also seen in the part of the tube farthest from interface: (1) 0.5%, 5 μm; (2) 1%, 3 μm; (3) 1.5%, 2.5 μm; (4) 2%, 2 μm. All the data indicate a decrease in the size of the particles as the gel is made thicker (Fig. 3D). This can be attributed to the decrease in pore size of the gel accompanying the increase in its thickness. Since agar gel acts as a scaffold in the synthesis process, decreasing the pore size of the gel yields particles of smaller size as the growth of crystals becomes more constrained by the gel thickness.5. Effect of temperature
The standard solution enthalpy of lanthanum hydroxide (ΔH0) was measure to be −151 kJ mol−1 with a solubility product Ksp0 2.5 × 10−22.44 Temperature was also tested to control the size of the spheres. Three temperatures were used (10 °C, 20 °C, 30 °C) while keeping the concentrations of inner and outer electrolytes constant at 50 mM and 14.8 M. The results are displayed in Fig. 3E which clearly exhibits a noticeable dependence of temperature: smaller spheres are obtained at higher temperatures which is counterintuitive but emphasizes the interplay between the kinetics of nucleation and growth, which increase with temperature, with the thermodynamics of dissolution, which decreases with temperature. An increase of 20 °C resulted in the decrease from 4 μm to 2 μm in the average size of the spheres in region (3), and a decrease from 0.7 μm to 0.4 μm in region (1).6. Particle size distribution
The mechanism of the spheres formation appears to be complex and dependent on several variables such as the concentration of electrolytes, temperature, and thickness of gel medium. As an attempt to understand the system, we take advantage of the stability of the fabricated La(OH)3 spheres in the agar gel matrix to investigate the spatial evolution of the size of the particles along the one-dimensional tube. For this purpose, we use scanning electron microscopy to monitor the growth of the spheres over nine consecutive locations in a tube prepared using 50 mM and 14.8 M as inner and outer electrolytes respectively in 1% agar matrix. The obtained results (Fig. 4) show a consistent increase in the average particle size from ∼350 nm near the interface separating the two electrolytes to an approximate size of ∼7.0 μm at the end of the tube. This observed space-dependent size evolution is in good agreement with the reaction–diffusion framework that exploits the supersaturation gradient along the tube. Near the interface, nucleation dominates and smaller particles are obtained; however down the tube, growth is more important than nucleation resulting in larger particles. In between, there is an Ostwald ripening mechanism where the small crystals that form in the beginning dissolve to reconstruct larger more crystalline particles. The proposed explanation is that the loosely-bound surface atoms first dissolve as an attempt to lower the system's energy, then as the solution gets supersaturated, the liberated atoms condense on the surface of growing particles, resulting in larger crystals.45,46 This gives rise to the bands47 that are observed in Fig. 1. It is also noticeable that the distribution of the size within a band or neighboring bands is narrow as shown by the red lines in Fig. 4. As a matter of fact, the particle size per band is almost uniform. | ||
| Fig. 4 Particle size distribution of the solid extracted from 9 different locations along the tube with the interface location as origin. [La3+] = 50 mM and [NH3] = 14 M. | ||
Agar – the organic matrix in the synthesis – seems essential for directing the growth of La(OH)3 crystals. This is evident when we observe how the use of another matrix such as gelatin results in a completely different structure of platelet-like morphology. To better understand the mechanism that is taking place in agar, we compare the SEM images of the precipitates near the interface, where we can observe the formation of the particles after nucleation, with the images of the particles away from the interface, where we can discern the grown particles. The self-assembled particles appear to emerge first in the form of platelets that aggregates before their transition into perfect spherules. Fig. 5 shows the outer surface of a spherule to be embedded by numerous cavities. A close look at a fractured La(OH)3 spherule in Fig. 6 shows that it is built by an organized packing of platelet substructures around a core that constitutes about 2–3% of the total spherical volume. Owing to the slow nucleation and aggregation in the gel matrix, it is made possible to study distinct intermediates of the progressive stages of crystal growth. The formation of large spherules is thought to begin when small platelets organize themselves into spherical structures, and the process is completed when several subunits combine and grow together into larger spherical structures.
 | ||
| Fig. 5 (A) SEM image of a typical spherical particle. (B) A close-up SEM image that illustrates the details of the topology of the spherical surface. [La3+] = 500 mM and [NH3] = 14 M. | ||
 | ||
| Fig. 6 SEM image of a broken spherical particle extracted away form the interface and revealing its interior structure. (B) A close-up SEM image displaying the cavity at the center of the sphere. (C) A close-up SEM image showing the aggregation of platelets as building blocks of the spherical microstructure. [La3+] = 500 mM and [NH3] = 14 M. | ||
E. Application-Congo red adsorption
Lanthanum hydroxide microspheres showed a very high adsorption capacity for Congo Red (CR) under optimal conditions. The spheres showed minor morphological changes after adsorption (ESI S4†). The pH was found to be a crucial parameter affecting the behavior of the dye molecule at the solid–liquid interface, where the best removal of CR from the simulated wastewater (25 ppm) was achieved at pH 5.0. 63% and 98% of the dye molecules were removed after 2 and 60 min, respectively, as shown in Fig. 7. Moreover, the size of the tested microspheres was another critical parameter that influences their adsorption capacities. Contrary to what is expected about the relation between spherical size and adsorption capacity, we find that for our spheres the bigger is the microsphere size, the higher is its CR removal capacity, which emphasizes the role of the complex surface topology and the role of the macroscopic pores on the adsorption of the dyes. Moreover, a crucial drop in the dye intake by the microspheres was observed after calcination at a temperature of 800 °C to give lanthanum oxide without noticeable change in the morphology of the spheres (ESI S5 and S6†). This suggests that the adsorption of the dye molecules is structure-dependent where surface hydroxyl groups (OH) play the key-role in monitoring the surface–dye interactions. The complete thermodynamic and kinetic studies conducted at different initial conditions showed that the experimental data fit the Langmuir isotherm (qmax = 330 mg g−1) and follow a pseudo-second order kinetic model. These exceptionally interesting results open the door for potential applications in the environmental field of lanthanum hydroxide microspheres obtained by reaction–diffusion of initially separated electrolytes within organic gels.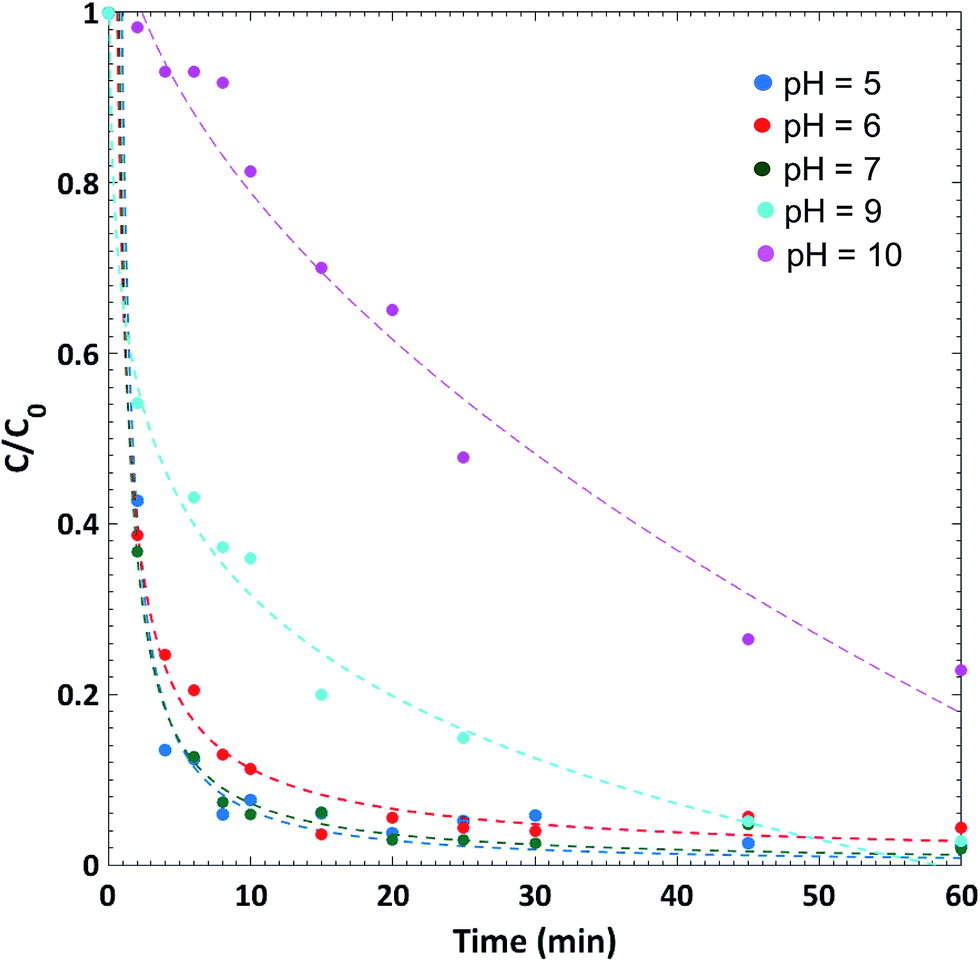 | ||
| Fig. 7 Kinetics of Congo Red (CR) dye removal at different pH. At optimal pH = 5, 95% of the dye is removed in about 8 minutes. [CR] = 25 ppm; volume of CR solution = 20 mL; mass of lanthanum hydroxide = 10 mg; temperature is 30 °C. | ||
F. Conclusion
We report a new synthetic route for the fabrication of La(OH)3 microspheres that is based on a simple low-cost reaction–diffusion framework. Our employed synthetic route can be implemented as well in the synthesis of hydroxides of other lanthanides of spherical morphology. To the best of our knowledge, this is the first report of a perfectly spherical morphology different from the typically synthesized rod-shaped particles. We also attempt to control the microscopic structure by controlling the concentration of the inner electrolyte and the thickness of the gel. In this regard, we take advantage of the spatial evolution of the particle size taking place as another method of controlling the spherical morphology to a selected size. The underlying mechanism appears to be complex but is shown to fit an Ostwald ripening model. Moreover, the detection of various intermediates including rods, dumbbells, and aggregates of subunits provides strong evidence for rod-dumbbell-sphere mechanism that is proposed for biomimetic crystallization using double-hydrophilic block copolymers. We hope our suggested synthetic route gives further insight to understand the complex bio-mineralization mechanisms. We also find it worthy to investigate the selective adsorption that lanthanum hydroxide material manifests on Congo red dye, along with the potential implications on waste-water treatment. The results of this study will be presented in another manuscript.Acknowledgements
The authors are grateful for the funding provided by the University Research Board at the American University of Beirut.Notes and references
- M. Fleys, W. Shan, Y. Simon and P.-M. Marquaire, Ind. Eng. Chem. Res., 2007, 46, 1063–1068 CrossRef CAS.
- H. Furuno, T. Hanamoto, Y. Sugimoto and J. Inanaga, Org. Lett., 1999, 2, 49–52 CrossRef.
- A. R. Strzelecki, P. A. Timinski, B. A. Helsel and P. A. Bianconi, J. Am. Chem. Soc., 1992, 114, 3159–3160 CrossRef CAS.
- N. Imanaka, K. Okamoto and G.-y. Adachi, Electrochem. Commun., 2001, 3, 49–51 CrossRef CAS.
- M. Ferhi, K. Horchani-Naifer and M. Férid, J. Lumin., 2008, 128, 1777–1782 CrossRef CAS.
- G. Yi, B. Sun, F. Yang, D. Chen, Y. Zhou and J. Cheng, Chem. Mater., 2002, 14, 2910–2914 CrossRef CAS.
- A. H. Peruski, L. H. Johnson Iii and L. F. Peruski Jr, J. Immunol. Methods, 2002, 263, 35–41 CrossRef CAS PubMed.
- A.-C. Texier, Y. Andrès, M. Illemassene and P. Le Cloirec, Environ. Sci. Technol., 2000, 34, 610–615 CrossRef CAS.
- V. Mahalingam, F. Vetrone, R. Naccache, A. Speghini and J. A. Capobianco, Adv. Mater., 2009, 21, 4025–4028 CrossRef CAS.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- A. A. Dakhel, Colloids Surf., A, 2009, 332, 9–12 CrossRef CAS.
- P. Schuetz and F. Caruso, Chem. Mater., 2002, 14, 4509–4516 CrossRef CAS.
- J. A. Capobianco, F. Vetrone, J. C. Boyer, A. Speghini and M. Bettinelli, Opt. Mater., 2002, 19, 259–268 CrossRef CAS.
- H. Liu, L. Wang, S. Chen and B. Zou, J. Lumin., 2007, 126, 459–463 CrossRef CAS.
- M. J. Escudero, X. R. Nóvoa, T. Rodrigo and L. Daza, J. Power Sources, 2002, 106, 196–205 CrossRef CAS.
- N. Imanaka, K. Okamoto and G.-y. Adachi, Angew. Chem., Int. Ed., 2002, 41, 3890–3892 CrossRef CAS.
- P. Pisecny, K. Husekova, K. Frohlich, L. Harmatha, J. Soltys, D. Machajdik, J. P. Espinos, M. Jergel and J. Jakabovic, Mater. Sci. Semicond. Process., 2004, 7, 231–236 CrossRef CAS.
- G. Azimi, R. Dhiman, H.-M. Kwon, A. T. Paxson and K. K. Varanasi, Nat. Mater., 2013, 12, 315–320 CrossRef CAS PubMed.
- J. Y. Li, H. Dai, X. H. Zhong, Y. F. Zhang, X. F. Ma, J. Meng and X. Q. Cao, J. Alloys Compd., 2008, 452, 406–409 CrossRef CAS.
- J. Sun, X.-P. Qiu, F. Wu and W.-T. Zhu, Int. J. Hydrogen Energy, 2005, 30, 437–445 CrossRef CAS.
- A. Tsubouchi and T. C. Bruice, J. Am. Chem. Soc., 1995, 117, 7399–7411 CrossRef CAS.
- Q. Mu and Y. Wang, J. Alloys Compd., 2011, 509, 396–401 CrossRef CAS.
- Z. Ji-Iing, Z. Yun-hong and Y. Hanxi, J. Power Sources, 1997, 69, 169–173 CrossRef.
- M. Zhou, J. Yuan, W. Yuan, Y. Yin and X. Hong, Nanotechnology, 2007, 18, 405704 CrossRef.
- C.-z. Yao, B.-H. Wei, H.-x. Ma, Q.-j. Gong, K.-w. Jing, H. Sun and L.-x. Meng, Mater. Lett., 2011, 65, 490–492 CrossRef CAS.
- S. Nielsen, J. J. Sloth and E. H. Hansen, Talanta, 1996, 43, 867–880 CrossRef CAS PubMed.
- X. Wang and Y. Li, Angew. Chem., Int. Ed., 2002, 41, 4790–4793 CrossRef CAS PubMed.
- N. Zhang, R. Yi, L. Zhou, G. Gao, R. Shi, G. Qiu and X. Liu, Mater. Chem. Phys., 2009, 114, 160–167 CrossRef CAS.
- B. Tang, J. Ge, C. Wu, L. Zhuo, J. Niu, Z. Chen, Z. Shi and Y. Dong, Nanotechnology, 2004, 15, 1273 CrossRef CAS.
- M. Salavati-Niasari, G. Hosseinzadeh and F. Davar, J. Alloys Compd., 2011, 509, 134–140 CrossRef CAS.
- Z. Liu, D. Zheng, Y. Su, Z. Liu and Y. Tong, Electrochem. Solid-State Lett., 2010, 13, E15–E18 CrossRef CAS.
- G. Li, C. Li, Z. Xu, Z. Cheng and J. Lin, CrystEngComm, 2010, 12, 4208–4216 RSC.
- M. Ozawa, R. Onoe and H. Kato, J. Alloys Compd., 2006, 408–412, 556–559 CrossRef CAS.
- I. Djerdj, G. Garnweitner, D. Sheng Su and M. Niederberger, J. Solid State Chem., 2007, 180, 2154–2165 CrossRef CAS.
- D. Zheng, J. Shi, X. Lu, C. Wang, Z. Liu, C. Liang, P. Liu and Y. Tong, CrystEngComm, 2010, 12, 4066–4070 RSC.
- C. G. Hu, H. Liu, W. T. Dong, Y. Y. Zhang, G. Bao, C. S. Lao and Z. L. Wang, Adv. Mater., 2007, 19, 470–474 CrossRef CAS.
- J. Zhu, Z. Gui and Y. Ding, Mater. Lett., 2008, 62, 2373–2376 CrossRef CAS.
- X. Wang, X. M. Sun, D. Yu, B. S. Zou and Y. Li, Adv. Mater., 2003, 15, 1442–1445 CrossRef CAS.
- P. Bocchetta, M. Santamaria and F. Di Quarto, Electrochem. Commun., 2007, 9, 683–688 CrossRef CAS.
- M. Mazloumi, S. Zanganeh, A. Kajbafvala, M. Shayegh and S. Sadrnezhaad, Nanosci. Nanotechnol., 2008, 4, 5 Search PubMed.
- M. Al-Ghoul, M. Ammar and R. O. Al-Kaysi, J. Phys. Chem. A, 2012, 116, 4427–4437 CrossRef CAS PubMed.
- J. Rahbani, M. Ammar and M. Al-Ghoul, J. Phys. Chem. A, 2013, 117, 1685–1691 CrossRef CAS PubMed.
- L. Ratke and P. W. Voorhees, Growth and coarsening: Ostwald ripening in material processing, Springer Science & Business Media, 2013 Search PubMed.
- L. R. Morss, Handbook of Physics and Chemistry of Rare Earths, 1994, 18, 239–291 Search PubMed.
- W. Ostwald, Z. Phys. Chem., 1897, 22, 289–330 CAS.
- P. W. Voorhees, J. Stat. Phys., 1985, 38, 231–252 CrossRef.
- Z. Shreif, M. Al-Ghoul and R. Sultan, ChemPhysChem, 2002, 3, 592–598 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra22692a |
| This journal is © The Royal Society of Chemistry 2016 |