
Luminescence and structural analysis of Ce3+ and Er3+ doped and Ce3+–Er3+ codoped Ca3Sc2Si3O12 garnets: influence of the doping concentration in the energy transfer processes
R. Fernández-Gonzálezac,
J. J. Velázquez*abd,
V. D. Rodríguezbc,
F. Rivera-Lópeze,
A. Lukowiakf,
A. Chiaserag,
M. Ferrarig,
R. R. Gonçalvesh,
J. Marrero-Jerezac,
F. Lahozbi and
P. Núñezac
aDepartamento de Química, U.D. Química Inorgánica, Universidad de La Laguna, 38206 La Laguna, Tenerife, Spain. E-mail: josvel@icv.csic.es
bDepartamento de Física, Universidad de La Laguna, 38206 La Laguna, Tenerife, Spain
cInstitute of Materials and Nanotechnology, Universidad de La Laguna, 38206 La Laguna, Tenerife, Spain
dInstituto de Cerámica y Vidrio (CSIC), C/Kelsen 5, 28049 Madrid, Spain
eSistema Multitécnicas de Análisis de Superficies, Servicio General de Apoyo a la Investigación (SEGAI), Universidad de La Laguna, 38206, La Laguna, Tenerife, Spain
fInstitute of Low Temperature and Structure Research PAS, 50-422 Wroclaw, Poland
gIFN-CNR CSMFO Lab., and FBK Photonics Unit, I-38123 Trento, Italy
hDepartamento de Química, Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto, Universidade de São Paulo, Av. Bandeirantes, 3900, CEP 14040-901, Ribeirão Preto, SP, Brazil
iInstituto Universitario de Estudios Avanzados en Atómica, Molecular y Fotónica (IUdEA), Universidad de La Laguna, 38206, La Laguna, Tenerife, Spain
First published on 26th January 2016
Abstract
We present research on Ca3Sc2Si3O12 garnets doped with Ce3+ and Er3+ ions that were synthesized by the freeze-drying precursor method. The structural characterization was performed by X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). Scanning electron microscopy (SEM) images of the calcined material were studied. Nanocrystals of Ca3Sc2Si3O12 were obtained by calcining the precursor at 1200 °C for 4 h with a mean size of about 100 nm. The luminescence features of Ce3+ and Er3+ doped samples indicated that these ions are effectively incorporated into the crystalline phase. In addition, codoping with Ce3+–Er3+ ions results in energy transfer processes from Ce3+ to Er3+ ions which led to the enhancement of near-infrared luminescence at 1.5 μm. The efficiency of this non-radiative energy transfer process has been studied by means of excited state dynamics as a function of the doping concentration. From these results we have found that near-infrared Er3+ luminescence efficiency under VIS Ce3+ excitation is at maximal for 1Ce3+–1Er3+ codoped Ca3Sc2Si3O12 garnets (in mol%). Luminescence quenching at higher Er3+ ion concentrations is observed.
1. Introduction
Rare earth (RE) doped luminescent materials have attracted much attention due to their potential applications in optoelectronic technology. In particular, Er3+ doped materials could be helpful in germanium based photovoltaic systems to make better use of the solar spectrum. They could enhance photon access in the near infrared (NIR) region where germanium cells show the highest conversion efficiency. The down-conversion involved processes are based on an energy transfer mechanism between RE pairs, starting with sensitizers that have strong absorption in the UV-blue region.1,2One of the most promising candidates for use as a sensitizer is the Ce3+ ion. Absorption of Ce3+ originates from the allowed electric-dipole transition from the 4f ground state to the 5d excited one, resulting in a high absorption cross section in the UV-blue spectral region.3 This absorption can be tuned by selection of the host material due to the strong dependence of the Ce3+ ion level diagram on the crystalline field, which also has appreciable shifts on the luminescence band.4,5 On the other hand, with respect to the typical option of Yb3+ ions as the luminescent center after energy transfer from the Ce3+ 5d level,6 Er3+ ions are an excellent option for energy conveyance from UV to NIR photons at 1550 nm, because they could involve the obtainment of three NIR photons after absorption of UV-blue photons,7 with a potential effective quantum efficiency close to 300%. The use of Ce3+ ions as sensitizers enhances the absorption in the UV-blue region, where Er3+ ions only present relatively weak and narrow absorption peaks.
The combination of Ce3+ and Er3+ ions with a suitable host would optimize the conversion of UV-VIS radiation into NIR. In this sense, nano-garnets with the general formula A3B2C3O12 have been widely studied after doping with RE ions due to their potential applications in optoelectronics. In particular, a silicate garnet with the composition Ca3Sc2Si3O12 (CSSO) has been synthesized by different methods such as solid-state reaction, microwave or sol–gel. Moreover their luminescence properties have been studied after doping with several lanthanides (Ce3+, Tb3+, Pr3+, Eu3+ or Nd3+).8–15 But, as far as we know, they have not been studied after codoping with Ce3+ and Er3+.
In this work, Ce3+ and Er3+ single and codoped Ca3Sc2Si3O12 garnets have been synthesized using a freeze-drying precursor route. Structural characterization by XRD and SEM techniques was performed to confirm the crystal structure of the CSSO garnet and to observe the microstructure of the sintered samples. Cerium ions could be present in the samples as Ce4+ ions, which are not luminescent. By this reason XPS was used for estimating the relative concentration of the two oxidation states of Ce (III and IV) on the surface of the samples. Finally, photoluminescence properties of single doped and codoped garnets and excite state dynamics in codoped samples have been studied in order to analyze the efficiency of the energy transfer mechanisms.
2. Experimental
2.1. Synthesis
Undoped, Ce3+–Er3+ single- and co-doped Ca3Sc2Si3O12 polycrystalline powders were synthesized by the freeze-drying precursor method,16–18 using CaCO3 (99%, Merck), Sc2O3 (99.9%, Aldrich), tetraethyl orthosilicate (C8H20O4Si) (98%, Aldrich), Ce(NO3)3·6H2O (99.99%, Aldrich) and Er(NO3)3·5H2O (99.9%, Aldrich) as reagents. Table 1 presents the different dopant concentrations that have been considered in this work. Hydrated metal nitrates were previously studied by thermogravimetric analysis to determine the cation content. Stoichiometric quantities of the reagents were individually dissolved in distilled water and then mixed together. Afterwards, ethylenediaminetetraacetic acid (EDTA) (99.7% Aldrich) was added as the complexing agent, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand to metal molar ratio, to prevent precipitation.
:1 ligand to metal molar ratio, to prevent precipitation.
| Sample | Ce3+ | Er3+ |
|---|---|---|
| CSSO | 0 | 0 |
| CSSO–1Ce | 1 | 0 |
| CSSO–4Ce | 4 | 0 |
| CSSO–0.5Er | 0 | 0.5 |
| CSSO–1Er | 0 | 1 |
| CSSO–1Ce0.5Er | 1 | 0.5 |
| CSSO–1Ce1Er | 1 | 1 |
| CSSO–1Ce2Er | 1 | 2 |
| CSSO–1Ce4Er | 1 | 4 |
The pH of the solution, which was initially acidic, was adjusted to around 7–8 by adding aqueous ammonia. Drops of the as-obtained solution were subsequently flash-frozen by pouring them into liquid nitrogen. In this way, each frozen drop retains the homogeneous cation distribution of the original solution. Then, the sample was freeze-dried in a HetoLyolab 3000 (Heto-Holten A/S) freeze-dryer apparatus for 3 days. The obtained highly hygroscopic amorphous precursor was immediately pre-calcined at 400 °C for 2 hours to prevent rehydration and to eliminate organic matter. Finally, the dry powders were pressed at 10 Tm in a hydrostatic press in order to obtain pellets which were calcined in alumina crucibles at 1200 °C for 4 hours.
2.2. Experimental setup
X-ray analyses (XRD) were carried out by using a Philips diffractometer equipped with an X’Celerator detector and a Cu anode (Cu Kα1, α2) in the 20–80 2θ diffraction range. The microstructure of the samples was studied by scanning electron microscopy (SEM) (Jeol LTD, mod. JSM-6300). All samples were covered with a thin film of silver to avoid charging problems and to obtain better image definition. The high-resolution X-ray photoelectron spectroscopy (XPS) spectrum was collected on an ESCALAB 250 spectrometer, using a monochromatized Al Kα X-ray radiation (hν = 1486.6 eV) with a spot size of 650 μm. The spectrometer energy calibration was performed using the Au 4f7/2 and Cu 2p3/2 photoelectron lines. The chemical shifts in the spectra were corrected with reference to the binding energy of the C 1s core level spectrum, corresponding to the carbon contamination layer at 284.6 eV. This peak arises from a minor quantity of contaminants and is generally accepted to be independent of the chemical state of the sample under investigation. The spectrum was collected in constant analyzer energy (CAE) mode, with pass energy of 20 eV and with an energy resolution of about 0.1 eV. For all the measurements, the pressure in the ultra-high vacuum analysis chamber was lower than 8 × 10−9 mbar, avoiding ejected photoelectron interaction with gas molecules. Laser Raman spectra were recorded on a Jobin Yvon/Labram HR spectrometer using 632.8 nm excitation from a He–Ne laser. Double beam Shimadzu UV-2450 Scan UV-visible spectrophotometer was used to record the reflectance spectra over a wavelength range of 200–800 nm. Emission and excitation spectra were recorded by using a 300 W xenon lamp with a double-grating monochromator (SPEX 1680). These spectra were collected by using a spectrometer (ANDOR Shamrock) equipped with an UV-VIS-NIR CCD camera (Newton CCD) and a NIR InGaAs or a single-grating monochromator (SPEX 1681) equipped with a photomultiplier tube (Hamamatsu R928). The spectral response of the set-up was taken into account. The luminescence decays in the nanosecond scale were measured by using a LifeSpec II spectrometer from Edinburgh Instruments. The samples were excited by 70 ps pulses with a 1 MHz repetition rate produced by a semiconductor laser operating at 405 nm. A multi-channel plate photomultiplier tube was used for the detection. The system was operated in time correlated single photon counting mode and had an instrumental response function (IRF) with a half maximum width of about 70 ps. The millisecond kinetics of luminescence were recorded using an optical parametrical oscillator (OPO) pumped by the 3rd harmonic of a YAG:Nd laser as the excitation source. The signal was detected with a digital storage oscilloscope controlled by a personal computer. The samples were excited by 6 ns pulses with a 20 Hz repetition rate. The time resolution of this setup was 20 ns.3. Results and discussion
The freeze-drying synthesis technique shows some advantages with reference to other precursor methods like co-precipitation or sol–gel method. The co-precipitation methods require that the cation solubility in the reaction mixture be very low to achieve nominal composition, while in the sol–gel method residual carbon from the complexing agent or from the gelation agent can remain, which do not decompose until reaching high temperatures. The amorphous product obtained using the freeze-drying method only contains nitrates which decompose at low temperatures. This method has been successfully used in previous works, obtaining pure phases and nanostructured materials at relatively low temperatures.16–18 For all of these reasons, we have used the freeze-drying synthesis method for studying the influence of doping concentration in the energy transfer processes of CSSO doped with Ce3+ and Er3+ ions.3.1. Structural characterization
 | (1) |
 | ||
| Fig. 1 XRD patterns of undoped, Ce3+ and Ce3+–Er3+ doped Ca3Sc2Si3O12 heat treated garnets; the main reflection planes are indicated. Standard peaks of Ca3Sc2Si3O12 (JCPDS file 72-1969) have been included for comparison purposes. | ||
| CSSO–1Ce | 110 nm |
| CSSO–1Ce1Er | 138 nm |
| CSSO–1Ce4Er | 88 nm |
Regarding the crystal structure of the CSSO garnets, it corresponds to the cubic system with Ia3![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) space group (#230).6 The Ca2+ ions are situated in the center of eight-coordinated dodecahedra (24c), while Sc3+ ions are located in the center of six coordinated octahedra (16a) and Si4+ ions are in the center of four-coordinated tetrahedra (24d). It is known that in the Ca3Sc2Si3O12 garnet structure one dodecahedron is associated to four other dodecahedra and four octahedral; two tetrahedra by shared edges and the other four octahedral by corner sharing.21 According to the literature, Ca2+ ions (ionic radius 114 pm) are substituted for Ce3+ and Er3+ ions in the dodecahedral position and do not substitute Sc3+ ions because of the difference of the respective ionic radius, 75 pm for Sc3+ and up to 100 pm for Ce3+ (115 pm) and Er3+ (103 pm).22–25
space group (#230).6 The Ca2+ ions are situated in the center of eight-coordinated dodecahedra (24c), while Sc3+ ions are located in the center of six coordinated octahedra (16a) and Si4+ ions are in the center of four-coordinated tetrahedra (24d). It is known that in the Ca3Sc2Si3O12 garnet structure one dodecahedron is associated to four other dodecahedra and four octahedral; two tetrahedra by shared edges and the other four octahedral by corner sharing.21 According to the literature, Ca2+ ions (ionic radius 114 pm) are substituted for Ce3+ and Er3+ ions in the dodecahedral position and do not substitute Sc3+ ions because of the difference of the respective ionic radius, 75 pm for Sc3+ and up to 100 pm for Ce3+ (115 pm) and Er3+ (103 pm).22–25
 | ||
| Fig. 2 SEM micrographs of 1Ce3+–xEr3+ (x = 0, 1, 2, 4), in mol%, co-doped Ca3Sc2Si3O12 garnets as a function of Er3+ ion concentration. | ||
The mean size of the aggregated grains, around 40 μm, is bigger than those obtained by other conventional synthesis processes like sol–gel combustion or emulsion-evaporation methods, in which higher heat treatment temperatures are necessary to obtain aggregated grains around 1–10 μm.26,27 On the other hand, it can be noted that the morphology of the samples seems to be affected by the Er3+ dopant concentration. When the Er3+ concentration increases from 0 to 1 mol%, an increment of the sinterization level could be observed. Further increases in the Er3+ concentration, from 2 to 4 mol%, does not result in a significant change of the sintering.
Fig. 3 shows the high resolution XPS spectra of Ce 3d in the binding energy (BE) range of 870–930 eV. The peaks between 870 and 895 eV belong to the Ce 3d5/2 levels while peaks between 895 and 913 eV correspond to the Ce 3d3/2 levels. The subscripts of U and V correspond to the oxidation state of Ce and the superscripts are an arbitrary label in order to differentiate the peaks of the same oxidation state.
 | ||
| Fig. 3 High resolution XPS spectra of the Ce 3d region (solid line) in a CSSO–4Ce garnet. Deconvolution of the peaks is indicated in dotted lines. The red dashed line represents the best fit obtained taking into account all deconvolutions. | ||
The satellite peak at around 917 eV, which is not observed in the initial state of trivalent Ce, is a characteristic fingerprint of the presence of cerium(IV), indicating that there is a mixture of Ce(III) and Ce(IV) ions in the sample.28
As it can been observed, the shape of the Ce 3d spectral line is complex due to the overlapping of several doublet lines resulting from different final states of cerium after the emission of photoelectrons. Thus, it is well accepted that the presence of Ce4+ ions leads to the appearance of three doublets and the Ce3+ ions produce two doublets in the XPS spectrum, with a total sum of ten lines. For the quantitative calculations of the Ce3+ and the Ce4+ concentrations, the high resolution XPS spectra of Ce 3d were deconvoluted in ten lines (see Fig. 3), according to the oxides of Ce(III)2O3 and Ce(IV)O2.29 In this work, U and V notations are used for the assignment of Ce 3d corresponding to 3d3/2 and 3d5/2, respectively.
In the Ce3+ species, V3+1–U3+1 are the shake down peaks and V3+2–U3+2 are the main peaks. In Ce4+, two shake down peaks V4+1–U4+1 and V4+2–U4+2 were assigned, and the main peaks V4+3–U4+3 were the characteristic peaks of Ce4+.
The concentration of Ce3+ and Ce4+ can be determined from the ratio of the integrated peak areas according to the following equations:30
 | (2) |
It could be said that XPS is a surface technique which explores materials until a depth of about 10 nm. However, when this technique is used, as in our case, with crystal sizes of about 100 nm, the analysed volume corresponds to about half of the total volume of the nanocrystals. Therefore, the XPS results indicate that a large fraction of the cerium ions is present in our sample as Ce3+ and then participate in the luminescence processes.
 | ||
| Fig. 4 Raman spectra of CSSO and CSSO–Ce obtained under 632.8 nm laser excitation. | ||
According to previous studies, Ca3Sc2Si3O12 garnets have 25 Raman active vibration modes expected on the basis of the analysis of the cubic space group Ia3 of the silicate garnets.21,31–35 In particular, 13 of them are observed in the region of 200–950 cm−1 as can be observed in Fig. 4, together with the peak at 420 cm−1 corresponding to the mode Fg of Sc2O3 phase.30 According to these studies, the band assignments can be summarized as follows.
The high frequency modes (800–950 cm−1) are related to symmetric and asymmetric internal stretching vibrations of rigid SiO4 tetrahedra. The modes lying from 450 to 700 cm−1 are assigned to bending motions of these tetrahedra. Finally, the remaining lattice modes (200–415 cm−1) involve rotations and translations of the SiO4 groups, octahedrally coordinated trivalent cations and dodecahedrally coordinated divalent cations. On the other hand, in a previous work we demonstrated that the intensity reduction of the peak corresponding to the Sc2O3 phase when doping with RE3+ ions is in agreement with the XRD results. Moreover, this Sc2O3 phase almost vanishes by RE3+ doping.19
3.2. Spectroscopy analysis
 | ||
| Fig. 5 Absorption spectra of Ce3+ and Er3+ doped and Ce3+–Er3+ codoped CSSO garnets. Spectra are multiplied by a factor of 20 in the range of 500–700 nm. | ||
3.2.2.1. Ce3+ and Er3+ single doped garnets. The photoluminescence spectra of Ce3+ and Er3+ 1 mol% single-doped CSSO garnets are shown in Fig. 6. The excitation and emission spectra corresponding to 1 mol% Ce3+ doped garnets are similar to those obtained by authors in a previous work.19 Moreover, these spectra are also similar to those obtained by Cheng et al.33 for Ce3+ ions in a closed matrix. The emission spectrum shows a broad emission band with two components, at 505 and 550 nm. The component at 505 nm corresponds to the interconfigurational transition of the Ce3+ ions from the lowest energy 5d level to the ground level, i.e. 5D3/2 → 2F5/2, shown in Fig. 6. The component located at 550 nm corresponds to the transition from the same emitting level to the first excited 4f level, i.e. 5D3/2 → 2F7/2. Moreover, the excitation spectrum of these emissions presents two peaks, at 311 and 450 nm. The peak at 450 nm is two orders of magnitude more intense.
 | ||
| Fig. 6 Normalized excitation and emission spectra of Ce3+ (black) and Er3+ (red) 1 mol% single-doped CSSO garnets with Ce3+ and Er3+, respectively. | ||
On the other hand, in the sample single-doped with Er3+ under excitation at 378 nm, that corresponds to the 4I15/2 → 4G11/2 transition of Er3+ ions, emissions at 545, 560, 660 and 850 nm are observed, see Fig. 6. These emissions correspond to transitions from the 2H11/2, 4S3/2, 4F9/2 and 4I9/2 levels to the ground state 4I15/2, respectively. These peaks, with well resolved Stark components, are typical of ions in crystalline environments and they are indicative of the effective incorporation of Er3+ ions into the CSSO garnets.
3.2.2.2. Ce3+–Er3+ codoped garnets. The incorporation of Er3+ ions into Ce3+–Er3+ codoped CSSO garnets results in a decrease of the Ce3+ emission intensity obtained under direct excitation of these ions, see Fig. 7. An excitation wavelength of 425 nm was selected in order to excite Ce3+ ions without simultaneous excitation of Er3+ ions. However, VIS and NIR luminescence of Er3+ ions at 540, 850 and 1550 nm is also observed, with the 1550 nm emission being much more intense than the others, as is shown in Fig. 7. It is noted that the highest emission intensity at 1550 nm is obtained for the Er3+ intermediate concentration of 1.0 mol%.
 | ||
| Fig. 7 (A) VIS-NIR emission spectra of Ce3+–Er3+ codoped CSSO garnets under excitation at 425 nm. Inset shows a magnified VIS spectrum of a 1Ce3+–4Er3+ codoped CSSO garnet. (B) Energy level diagrams of Ce3+ and Er3+ ions with main excitation and emission transitions, indicated by solid arrows, and energy transfer mechanisms, indicated by dashed arrows. | ||
These Er3+ emissions are due to an energy transfer from Ce3+ to Er3+ ions. These energy transfer processes are caused by the overlap of the broad Ce3+ emission at 505–550 nm, see Fig. 6, with the Er3+ absorption, mainly at 520 nm (4I15/2 → 2H11/2), see Fig. 5.
On the other hand, the decrease of the Ce3+ emission with the increase of the Er3+ concentration is not followed by an equivalent enhancement of the NIR emission of these ions, as observed in Fig. 7. For this reason, the excited state dynamics after pulsed excitation were studied in order to analyse the mechanisms involved in the luminescence of these materials.
 | (3) |
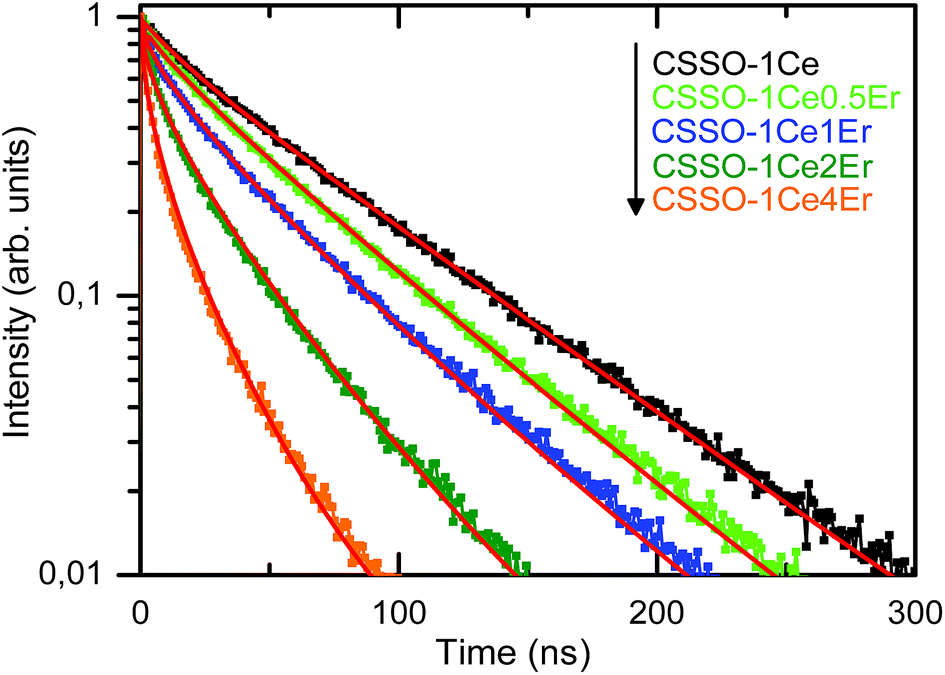 | ||
| Fig. 8 Decays of Ce3+ doped and Ce3+–Er3+ codoped CSSO garnets with excitation at 405 nm and detection at 505 nm. The red lines represent the best fits of the experimental results to eqn (3). | ||
On the other hand, the energy transfer efficiency from Ce3+ to Er3+ ions, ηET, can be calculated by using eqn (4),
 | (4) |
| Samples | τCe-xEr (ns) | ηET | τxEr (ms) | ηEr-emis |
|
|
|---|---|---|---|---|---|---|
| Calc. | Exp. | |||||
| CSSO–1Ce | 56.0 | — | — | — | ||
| CSSO–1Ce0.5Er | 44.5 | 0.21 | 6.0 | 1.0 | 0.27 | 0.10 |
| CSSO–1Ce1Er | 33.6 | 0.40 | 5.9 | 0.98 | 0.65 | 0.32 |
| CSSO–1Ce2Er | 19.8 | 0.65 | 1.6 | 0.27 | 0.50 | 0.28 |
| CSSO–1Ce4Er | 10.0 | 0.82 | 0.9 | 0.15 | 0.68 | 0.45 |

Having such hints, we measured the decay of the NIR Er3+ emission at 1550 nm after pulsed excitation at 405 nm, corresponding to direct excitation of Er3+ ions. These decays, shown in Fig. 9, present a single exponential behaviour for the lowest Er3+ concentrations of 0.5 and 1 mol%, with a lifetime of 6.0 ms. Meanwhile, for concentrations above 1 mol% the emission decays are quite faster, with effective lifetimes presented in Table 3. This behaviour would be due to quenching of the emission by energy migration until luminescence is trapped.
 | ||
| Fig. 9 Decays of Ce3+–Er3+ codoped CSSO garnets excited at 405 nm and detected at 1550 nm, corresponding to the 4I13/2 → 4I15/2 transition. | ||
From these effective lifetimes, we obtained the quantum efficiency of the NIR Er3+ emission by using eqn (5):
 | (5) |
Now we can calculate the expected ratio of Er3+ to Ce3+ integrated emissions, taking into account the energy transfer efficiency from the Ce3+ ions and the quantum efficiency of the NIR Er3+ emission, by using eqn (6).
 | (6) |
The obtained results are shown in Table 3 and would be compared with the experimental values obtained directly from the integrated photon fluxes of Er3+ and Ce3+ emissions, from spectra in Fig. 7.
The experimental emission ratios obtained experimentally from spectra in Fig. 7 are lower than the values calculated by using eqn (5). This indicates that in Ce3+–Er3+ co-doped garnets the energy transfer from Ce3+ to Er3+ is not the only mechanism involved in the reduction of Ce3+ emission. A similar result was obtained in garnets codoped with Ce3+ and Tb3+ ions.19 But in this case we obtain a relatively high efficiency in the transfer from Ce3+ ions to acceptor ions. For the optimum composition of 1 mol% of Er3+, following Fig. 6, about half of the non-radiative decays of Ce3+ ions take to the NIR emission of Er3+ ions (from the comparison of experimental and calculated ratios IEr/ICe in Table 3).
4. Conclusions
Ca3Sc2Si3O12 garnets doped with Ce3+ and Er3+ ions have been successfully synthesized by a freeze-drying precursor method. After heat treatment at 1200 °C, Ca3Sc2Si3O12 nanocrystals, with a mean size of about 100 nm have been obtained. From XPS measurements we found that a large fraction of the cerium ions are present as Ce3+ ions in these garnets. Moreover, luminescence features indicated that Ce3+ and Er3+ dopant ions are effectively incorporated into the garnets. In addition, codoping with Ce3+–Er3+ ions results in energy transfer processes from Ce3+ to Er3+ ions, allowing for efficient obtainment of NIR emission at 1.5 μm under blue excitation. The efficiency of this non-radiative energy transfer process has been analyzed by means of excited state dynamics as a function of the doping concentration. From these results we found that NIR luminescence of the garnets is maximal for CSSO–C1E. For higher Er3+ concentrations, the energy transfer efficiency increases, up to 82% for 4 mol% of Er3+, but also an appreciable increase of quenching effects is observed for Er3+ concentrations over 1 mol%.Acknowledgements
The authors would like to thank Ministerio de Economía y Competitividad (MAT2013-42407-R cofunded by FEDER funds) and Fundación CajaCanarias (Proyecto HIDROSOLAR, ENER02) for financial support. J. J. Velázquez also thanks Ministerio de Economía y Competitividad for Posdoctoral grant (FPDI-2013-16895). Fernando Rivera-López thanks Cajasiete for financial support related with Servicio de Sistema Multitécnicas de Análisis de Superficies (SEGAI-ULL). We also thank the group MATMOL, from University of La Laguna (ULL), for the possibility of using the UV-Visible absorbance spectrophotometer. This research was partially performed in the framework of the projects CNR-PAS (2014–2016) and COST ACTION MP1401. R. R. Gonçalves and M. Ferrari acknowledge Brazilian Scientific Mobility Program “Ciências sem Fronteiras”.References
- B. M. van der Ende, L. Aarts and A. Meijerink, Phys. Chem. Chem. Phys., 2009, 11, 11081, 10.1039/b913877c.
- J. Zhou, Y. Teng, X. Liu, S. Ye, Z. Ma and J. Qiu, Phys. Chem. Chem. Phys., 2010, 12, 13759, 10.1039/c0cp00204f.
- F. Piccinelli, A. Speghini, G. Mariotto, L. Bovo and M. Bettinelli, J. Rare Earths, 2009, 27, 555, DOI:10.1016/s1002-0721(08)60287-2.
- D. Q. Chen, Y. S. Wang, Y. L. Yu, P. Huamg and F. Y. Weng, J. Appl. Phys., 2008, 104, 116105, DOI:10.1063/1.3040005.
- P. Dorenbos, J. Lumin., 2000, 91, 106, DOI:10.1016/s0022-2313(00)00197-6.
- J. Ueda and S. Tanabe, J. Appl. Phys., 2009, 106, 043101, DOI:10.1063/1.3194310.
- X. Chen, J. Wu, X. Xu, Y. Zhang, N. Sawanobori, C. Zhang, Q. Pan and G. J. Salamo, Opt. Lett., 2009, 34, 887, DOI:10.1364/ol.34.000887.
- Y. Suzuki, M. Kakihana, Y. Shimomura and N. Kijima, J. Jpn. Soc. Powder Powder Metall., 2007, 54, 44, DOI:10.2497/jjspm.54.44.
- Y. Shimomura, T. Honma, M. Shigeiwa, T. Akai, K. Okamoto and N. Kijima, J. Electrochem. Soc., 2007, 154, 35, DOI:10.1149/1.2388856.
- K. V. Ivanovskikh, A. Meijerink, F. Piccinelli, A. Speghini, E. I. Zinin, C. Ronda and M. Bettinelli, J. Lumin., 2010, 130, 893, DOI:10.1016/j.jlumin.2009.12.031.
- M. Bettinelli, A. Speghini, F. Piccinelli, A. N. C. Neto and O. L. Malta, J. Lumin., 2011, 131, 1026, DOI:10.1016/j.jlumin.2011.01.016.
- C. Feng, Y. Meng, X. Liu, J. Wang, L. Yang and X. Jinshu, Chin. J. Rare Met., 2013, 87 DOI:10.3969/j.issn.0258-7076.2013.01.016.
- Y. Chen, J. Feng, Y. Pan, J. Li, S. Zeng, M. Liang, Z. Liu, N. Li and Y. Su, J. Lumin., 2014, 148, 153, DOI:10.1016/j.jlumin.2013.12.015.
- J. Qiao, J. Zhang, X. Zhang, Z. Hao, Y. Liu and Y. Luo, Dalton Trans., 2014, 43, 4164, 10.1039/c3dt52902a.
- W. Wan, Q. Xiao, C. Yang and K. Meng, J. Chin. Ceram. Soc., 2010, 38(10), 1862–1866 Search PubMed.
- M. O. Mazan, J. Marrero-Jerez, A. Soldati, P. Núñez and S. Larrondo, Int. J. Hydrogen Energy, 2015, 40, 3981, DOI:10.1016/j.ijhydene.2015.01.006.
- M. Amsif, D. Marrero-López, A. Magrasó, J. Peña-Martínez, J. C. Ruiz-Morales and P. Núñez, J. Eur. Ceram. Soc., 2009, 29, 131, DOI:10.1016/j.jeurceramsoc.2008.06.001.
- J. Marrero-Jerez, S. Larrondo, E. Rodríguez-Castellón and P. Núñez, Ceram. Int., 2014, 40, 6807, DOI:10.1016/j.ceramint.2013.11.143.
- J. J. Velázquez, R. Fernández-González, J. Marrero-Jerez, V. D. Rodríguez, A. Lukowiak, A. Chiappini, A. Chiasera, M. Ferrari and P. Núñez, Opt. Mater., 2015, 46, 109, DOI:10.1016/j.optmat.2015.03.057.
- B. D. Cullity, Elements of X-ray Diffraction, Addison-Wesley, Massachusetts, 1978 Search PubMed.
- W. Yun-Fang, C. Ya-Han, N. Yung-Tang and C. In-Gann, J. Am. Ceram. Soc., 2013, 96, 234, DOI:10.1111/jace.12034.
- Y. Liu, X. Zhang, Z. Hao, Y. Luo, X. Wang and J. Zhang, J. Mater. Chem., 2011, 21, 16379 RSC.
- M. S. Kishore, N. P. Kumar, R. G. Chandran and A. A. Setlur, Electrochem. Solid-State Lett., 2010, 13(6), J77–J80 CrossRef CAS.
- R. G. Burns, Ionic radii of transition metals and related cations, in Mineralogical Applications of Crystal Field Theory. Cambridge Topics in Mineral Physics and Chemistry. (No 5), Cambridge University Press, Cambridge, 2nd edn, 1993, pp. 464–465, DOI:10.1017/cbo9780511524899.016.
- V. Zepf, Rare Earth Elements. A New Approach to the Nexus of Supply, Demand and Use: Exemplified along the Use of Neodymium in Permanent Magnets, Springer Theses, Augsburg University, 2013, ISBN 978-3-642-35458-8.
- Y. Liu, J. Hao, W. Zhuang and Y. Hu, J. Phys. D: Appl. Phys., 2009, 42, 245102, DOI:10.1088/0022-3727/42/24/245102.
- N. Enomoto, T. Sakai, M. Inada, Y. Tanaka and J. Hojo, J. Ceram. Soc. Jpn., 2010, 118, 1067, DOI:10.2109/jcersj2.118.1067.
- E. Bêche, P. Charvin, D. Perarnau, S. Abanades and G. Flamant, Surf. Interface Anal., 2008, 40, 264, DOI:10.1002/sia.2686.
- S. Gavarini, M. J. Guittet, P. Trocellier, M. Gauthier-Soyer, F. Carrot and G. Matzen, J. Nucl. Mater., 2003, 322, 111, DOI:10.1016/s0022-3115(03)00306-4.
- J. Zhang, X. Ju, Z. Y. Wu, T. Liu, T. D. Hu, Y. N. Xie and Z. L. Zhang, Chem. Mater., 2001, 13, 4192, DOI:10.1021/cm010235p.
- N. D. Todorov, M. V. Abrashev, V. Marinova, M. Kadiyski, L. Dimowa and E. Faulques, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 104301 CrossRef.
- A. Ubaldini and M. M. Carnasciali, J. Alloys Compd., 2008, 454, 374 CrossRef CAS.
- Y. Cheng, K. Cheah and M. Gong, J. Lumin., 2011, 131, 1589 CrossRef.
- A. M. Hofmeister and A. Chopelas, Phys. Chem. Miner., 1991, 17, 503 CrossRef CAS.
- S. P. Feofilov, D. V. Arsentyev, A. B. Kulinkin and R. I. Zakharchenya, J. Lumin., 2011, 131, 438 CrossRef CAS.
- M. Inokuti and F. Hirayama, J. Chem. Phys., 1965, 43, 1978, DOI:10.1063/1.1697063.
- G. Zhang, C. Liu, J. Wang, X. Kuang and Q. Su, J. Mater. Chem., 2012, 22, 2226, 10.1039/c1jm14942c.
| This journal is © The Royal Society of Chemistry 2016 |