
Phenylselanyl-1H-1,2,3-triazole-4-carbonitriles: synthesis, antioxidant properties and use as precursors to highly functionalized tetrazoles†
Lucielli Savegnago*a,
Manoela do Sacramentob,
Lucimar M. P. Broda,
Mariana G. Fronzaa,
Natália Seusb,
Eder J. Lenardãob,
Márcio W. Paixãoc and
Diego Alves*a
aGrupo de Pesquisa em Neurobiotecnologia – GPN, CDTec, Universidade Federal de Pelotas, UFPel, Pelotas, RS, Brazil. E-mail: diego.alves@ufpel.edu.br
bLaboratório de Síntese Orgânica Limpa – LASOL – CCQFA – Universidade Federal de Pelotas – UFPel, CEP 96010-900, Pelotas, RS, Brazil. Tel: +55 (53) 3275-7357
cLaboratório de Síntese de Produtos Naturais, Universidade Federal de São Carlos, São Carlos 13565-905, SP, Brazil
First published on 15th December 2015
Abstract
We describe herein our results on the synthesis, antioxidant properties and chemical diversification of phenylselanyl-1H-1,2,3-triazole-4-carbonitriles. These compounds were synthesized in high yields by the reaction of azidophenyl phenylselenides with a range of α-keto nitriles, using DMSO as the solvent in the presence of a catalytic amount of Et2NH (1 mol%). The synthesized compounds were screened for their in vitro antioxidant activity and 5-phenyl-1-(2-(phenylselanyl)phenyl)-1H-1,2,3-triazole-4-carbonitrile (3a) exhibited the highest antioxidant effect. In addition, the obtained triazoyl carbonitriles were readily transformed into more complex products via a cycloaddition protocol with NaN3, affording bifunctional hybrids containing triazole and tetrazole systems in good to excellent yields.
Introduction
Heterocyclic systems have been pointed out as one of the most representative chemical architectures found in several natural and synthetic bioactive compounds.1 Among them, those containing nitrogen atoms are widely applied in chemical biology.2 In particular, 1,2,3-triazoles3 are an interesting class of N-heterocycles broadly used in drug-discovery and modulation of drug candidates.4 Several methods for the synthesis of this heterocycle have been already reported via 1,3-dipolar cycloaddition between azides and alkynes under both thermal5 and transition metal catalysis based on copper and ruthenium salts.6 However, the necessity for transition metals has restricted the application of these methodologies in chemical biology.7 For example, copper has been associated with cellular toxicity, metabolic disruption and Cu-induced oxidative damage in biological systems.8 To overcome this limitation, organocatalytic approaches involving β-enamine-azide or enolate-azide cycloadditions have been employed to promote the construction of functionalized 1,2,3-triazoles.9 Under different reaction conditions, the carbonyl compound could easily generate an enamine or an enolate – both species act as dipolarophiles in organocatalyzed 1,3-dipolar cycloadditions with organic azides.9aRecently, our research group described the application of β-enamine-azide cycloadditions for the synthesis of 1,2,3-triazoles bearing organoselenium moieties.10 A range of selanyl-triazoyl carboxylates and carboxamides were synthesized in good to excellent yields using catalytic amounts of Et2NH. Organoselenium compounds are valuable molecules in organic synthesis, since these scaffolds are important units in biological sciences11 and also serve as versatile building blocks in organic synthesis.12 Among them, those containing nitrogen atoms in their structures are a special class of molecules and have been used for several purposes.12 Selenium-containing 1,2,3-triazole compounds13 are an interesting and yet unexplored class of molecules that might have broader biological applications. These hybrid molecules combine the well-known activity of the 1,2,3-triazole ring3,4 with that of the selenium-containing group.12 For example, the synthetic 4-phenyl-1-(phenylselanylmethyl)-1,2,3-triazole (Se-TZ) demonstrated an antidepressant-like effect mediated, at least partially, via the central dopaminergic and serotoninergic neurotransmitter systems.14
As a continuation of our studies toward the development of new 1,2,3-triazoles bearing organoselenium moieties, we report herein the full results of the synthesis of phenylselanyl-1H-1,2,3-triazole-4-carbonitriles (3). The obtained compounds were screened for their in vitro antioxidant activity. In addition, these compounds were readily transformed into more complex structures by a second cycloaddition with NaN3, furnishing the desired bifunctional hybrids containing triazole and tetrazole rings (4, Fig. 1).
 | ||
| Fig. 1 General scheme of this work. | ||
Results and discussion
During our studies on the synthesis of arylselanyl-1H-1,2,3-triazole-4-carboxylates starting from azidophenyl arylselenides and β-keto-esters, it was observed that the selanyl-triazoyl carbonitrile (3a) could be efficiently obtained starting from 2-azidophenyl phenylselenide (1a) and using benzoylacetonitrile (2a) instead id β-keto-esters (Scheme 1).10a The reaction was performed in the presence of 10 mol% of Et2NH as the catalyst and DMSO as the solvent and, after 18 h at room temperature, the desired product 3a was obtained in 91% yield. | ||
| Scheme 1 Previous work. | ||
Owing to our interest in the synthesis of Se-containing triazoles, we decided to perform further control experiments looking for a general method to prepare densely functionalized compounds like 3a. To our delight, we observed that by decreasing the amount of Et2NH from 10 to 1 mol%, the desired product 3a was obtained in 95% yield after 1 h of reaction at room temperature (Table 1, entry 1). With this result in hand, we focused on extending the scope of this methodology by reacting 2-azidophenyl phenylselenide (1a) with a range of substituted α-keto nitriles under the optimized reaction conditions.
|
|||
|---|---|---|---|
| Entry | α-Keto nitriles 2a–g | Time (h) | Product (yield)b |
| a Reactions were performed with 2-azidophenyl phenylselenide 1a (0.33 mmol) and α-keto nitriles 2a–g (0.3 mmol) using DMSO as the solvent (0.3 mL) at room temperature under air atmosphere and were monitored by TLC until total disappearance of the starting materials.b Yields are given for isolated products.c Reaction performed at 70 °C gave the product 3g in 25% yield. | |||
| 1 |  |
1 | 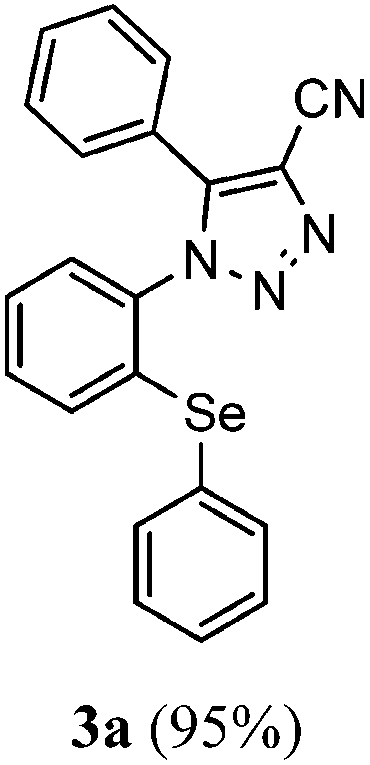 |
| 2 |  |
1 |  |
| 3 |  |
1 |  |
| 4 |  |
1 |  |
| 5 |  |
1.5 |  |
| 6 |  |
1 | 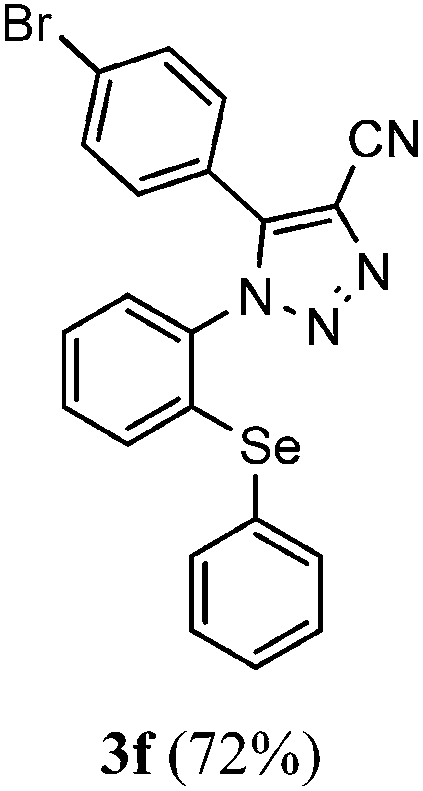 |
| 7 |  |
24 |  |

The results shown in Table 1 demonstrate that our protocol worked well for a range of substituted α-keto nitriles, affording good yields of the desired products. In general, the reactions were not sensitive to the electronic effect at the aromatic ring in the α-keto nitriles (2). Consequently, α-keto nitriles containing an electron-donating group (4-Me and 4-OMe) and an electron-withdrawing group (4-F, 4-Cl and 4-Br) at the aromatic ring delivered the desired selanyl-triazoyl carbonitriles 3b–f in good yields (Table 1, entries 1–6). Unfortunately, when the reaction was carried out with 4,4-dimethyl-3-oxopentanenitrile (2g), the corresponding product 3g was obtained in low yield, even running the reaction at 70 °C for 24 h (Table 1, entry 7).
We next evaluated the reactivity of 4-azidophenyl phenylselenide (1b) with benzoylacetonitrile (2a) under the same reaction conditions (Scheme 2). In this reaction, however, a mixture of regioisomers (3h and 3h′) was obtained in 90% yield after 2 h [ratio 3h/3h′ (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)].
:1)].
 | ||
| Scheme 2 Cycloaddition reaction between 1b and benzoylacetonitrile (2a). | ||
All the synthesized selanyl-triazoyl carbonitriles 3a–h were characterized by their mass, 1H and 13C NMR spectra (see ESI†).
The excessive production of reactive species by cellular respiration and other metabolic activities can cause damage to all cellular structures.15 Oxidative stress is critical to the etiology of many chronic and degenerative diseases, such as cancer, cardiovascular diseases, diabetes and obesity.16 In this sense, the search for new synthetic compounds with antioxidant potential has increased in recent years.17 Aiming to access the possible antioxidant effect of the new selanyl-triazoyl carbonitriles (3), they were subjected to different in vitro assays.
Iron is essential for life in many organisms and for normal neurological function; however, it can participate in redox reactions (electron transfer reactions) with a variety of substrates,18 which makes it the most important inducer of reactive species in the human organism.19 Based on this consideration, the ferric ion (Fe3+) reducing antioxidant power (FRAP) assay was performed to access the electron donation capability of the synthesized triazoles 3a–e.20 The FRAP of a compound is a valuable indicator of its potential antioxidant activity. It was observed that compound 3a exhibited a high ferric-reducing ability, which increased with its concentration (Table 2). Compounds 3b–e, on the other hand, did not show a significant effect at all the tested concentrations. Based on these data, compound 3a was the best ferric ion reductant.
| Absorbance at 593 nm | |
|---|---|
| a Data are presented as the mean ± S.E.M; (**) p < 0.01; (***) p < 0.001 as compared to the respective control sample (FRAP solution without compounds) (one way ANOVA/Newman–Keuls). | |
| Control | 0.37 ± 0.08 |
| Compound 3a (μM) | |
|---|---|
| 10 | 0.44 ± 0.05 |
| 50 | 0.73 ± 0.03** |
| 100 | 1.03 ± 0.03*** |
We also evaluated the capacity of the compounds 3a–e for neutralizing the reactive species (RS) in rat hippocampus (Table 3). The generation of RS can be induced by a variety of oxidants, one of which is azide, which acts by inhibiting the electron transport in Complex IV.21
| Compounds (μM) | 3a | 3b | 3d | 3e |
|---|---|---|---|---|
| 10 | 48.48 ± 25.94** | 50.12 ± 21.52* | 55.64 ± 15.53 | 54.04 ± 18.66* |
| 50 | 24.64 ± 10.54** | 29.51 ± 12.97** | 22.70 ± 6.26*** | 42.03 ± 17.54* |
| 100 | 16.84 ± 4.03** | 35.53 ± 14.84** | 17.89 ± 7.21*** | 37.48 ± 20.85* |
| 500 | 10.63 ± 2.66** | 15.58 ± 6.15** | 11.58 ± 3.44*** | 18.12 ± 4.86* |
| Imaxc (%) | 89.37 ± 4.61 | 84.41 ± 10.66 | 88.42 ± 5.96 | 81.95 ± 8.49 |
The results from the RS assay suggest that compounds 3a, 3b and 3e decreased the formation of reactive species induced by azide in the hippocampus at concentrations equal to or higher than 10 μM, while 3d was effective only from 50 μM (Table 3). The triazole 3c, containing the methoxyl group, did not show a significant effect. The maximum inhibition (Imax) observed was about 85%, which indicates the high efficacy of compounds 3a–b and 3d–e in this assay.
Compound 3c showed significant activity against RS in the cortex, drastically reducing their activity at the concentration of 500 μM (Table 4). Compounds 3b, 3d and 3e were able to decrease the formation of RS starting at the concentration of 50 μM, while 3a was effective at 10 μM. The Imax of the tested compounds in inhibiting RS in the cortex was around 78%. Based on these outcomes for the RS in the hippocampus and the cortex, it is not possible to infer a clear structure–activity relationship, once all compounds showed a significant ability to protect from the formation of RS. It is possible, in a bigger picture, that this new class of compounds could prevent the oxidative stress.
| Concentration (μM)/compound | 3a | 3b | 3c | 3d | 3e |
|---|---|---|---|---|---|
| 10 | 59.85 ± 21.52* | 69.87 ± 17.92 | 85.89 ± 7.63 | 92.03 ± 4.57 | 64.41 ± 19.60 |
| 50 | 40.12 ± 12.05* | 39.82 ± 10.50** | 56.44 ± 22.83 | 49.98 ± 12.36** | 40.39 ± 11.98* |
| 100 | 33.36 ± 11.63* | 35.89 ± 16.46** | 59.56 ± 13.92 | 40.39 ± 8.84*** | 39.85 ± 14.34* |
| 500 | 24.81 ± 11.34** | 12.94 ± 6.40** | 23.28 ± 3.28** | 27.68 ± 7.29*** | 20.66 ± 11.08** |
| Imaxc (%) | 75.18 ± 19.63 | 87.26 ± 10.85 | 76.72 ± 5.68 | 72.32 ± 12.63 | 79.33 ± 19.19 |
Compounds 3a–e were evaluated for their capacity to reduce the sodium nitroprusside (SNP)-induced linoleic acid peroxidation. The photodegradation of SNP produces NO, which is a free radical with short half-life (<30 s) and when coupled with other ROS may cause neuronal damage.22 In this assay, all compounds reduced the sodium nitroprusside (SNP)-induced linoleic acid peroxidation (Table 5). Compounds 3a and 3e were effective at a concentration equal or superior to 10 μM, while 3b inhibits the lipid peroxidation from 50 μM. Compounds 3c and 3d showed significant effect only at higher concentrations (500 and 100 μM, respectively). The values of Imax were about 65% in this assay.
| Concentration (μM)/compound | 3a | 3b | 3c | 3d | 3e |
|---|---|---|---|---|---|
| 10 | 47.84 ± 18.15 | 74.50 ± 10.92 | 77.96 ± 12.14 | 79.03 ± 7.49 | 71.67 ± 13.08* |
| 50 | 40.68 ± 17.73* | 59.56 ± 14.28* | 74.75 ± 8.74 | 78.56 ± 9.96 | 58.21 ± 15.86* |
| 100 | 45.12 ± 6.79* | 49.10 ± 14.33* | 74.96 ± 10.78 | 44.36 ± 7.51** | 43.26 ± 13.42** |
| 500 | 35.79 ± 9.42* | 46.97 ± 11.86** | 56.91 ± 12.26* | 46.84 ± 14.22** | 48.54 ± 9.48** |
| Imaxc (%) | 69.01 ± 16.81 | 67.06 ± 13.60 | 64.53 ± 12.61 | 65.60 ± 14.96 | 65.72 ± 11.41 |
Due to the ability of compounds 3a–e to protect from the oxidation of linoleic acid, which is a lipid matrix, their potential for inhibiting the lipid peroxidation in the cortex and the hippocampus of rats was evaluated (Tables 6 and 7). It is known that lipid peroxidation in these brain regions is related with neurodegenerative disease, such as Alzheimer disease.23 The levels of TBARS after lipid peroxidation induced by SNP in the hippocampus were reduced for all compounds at different concentrations. As shown in Table 6, compound 3a was effective starting from a concentration of 50 μM, while 3b showed significant inhibition of the lipid peroxidation at concentrations of 10, 50 and 100 μM. Compounds 3c and 3d were able to protect against lipid peroxidation at 10 μM and 3e was effective only at 500 μM. Therefore, the Imax effect in the hippocampus was in the following order 3a ≥ 3d ≥ 3c ≥ 3b > 3e.
| Concentration (μM)/compound | 3a | 3b | 3c | 3d | 3e |
|---|---|---|---|---|---|
| Controlb | 47.17 ± 8.08 | 53.28 ± 7.90 | 47.56 ± 7.71 | 50.20 ± 12.49 | 46.57 ± 9.22 |
| 10 | 87.13 ± 12.87 | 61.28 ± 12.48** | 46.26 ± 10.49* | 54.28 ± 11.31* | 80.38 ± 5.18 |
| 50 | 56.90 ± 8.95* | 55.42 ± 5.85* | 75.28 ± 15.51 | 70.86 ± 8.29 | 75.12 ± 6.60 |
| 100 | 63.30 ± 8.37* | 56.90 ± 12.88* | 61.80 ± 5.87 | 59.08 ± 14.66 | 86.21 ± 7.94 |
| 500 | 47.54 ± 9.70** | 98.54 ± 8.72 | 72.14 ± 13.94 | 91.38 ± 8.62 | 62.64 ± 10.15* |
| Imaxc (%) | 61.46 ± 5.20 | 40.00 ± 17.84 | 50.52 ± 16.16 | 52.09 ± 19.48 | 39.55 ± 17.94 |
| Compounds (μM) | 3a | 3c | 3d |
|---|---|---|---|
| Controlb | 39.08 ± 4.36 | 49.77 ± 9.82 | 47.47 ± 10.79 |
| 10 | 76.50 ± 11.82 | 62.03 ± 5.49** | 60.50 ± 7.65* |
| 50 | 56.48 ± 22.10* | 62.58 ± 4.42** | 67.47 ± 7.78* |
| 100 | 31.67 ± 8.06** | 53.61 ± 8.03** | 66.98 ± 11.07* |
| 500 | 16.47 ± 2.43** | 78.68 ± 9.07* | 72.76 ± 6.86* |
| Imaxc (%) | 83.53 ± 4.21 | 53.18 ± 9.02 | 42.63 ± 16.60 |
Furthermore, in the cortex, compound 3a was effective starting at the concentration of 50 μM, while compounds 3c and 3d were able to protect against lipid peroxidation at concentrations equal or higher than 10 μM, with an Imax of 3a > 3c ≥ 3d (Table 7). The compound 3a was the most effective in protect the lipid peroxidation among the other in hippocampus and cortex of rats.
In view of the results presented above, compound 3a exhibited a better antioxidant effect when compared to compounds 3b–e, owing to higher value of Imax, and so greater efficacy. It is worth mentioning that this compound has no substituents in their structure leading us to believe that the presence of electron donating groups (–CH3, –OCH3) or electron-withdrawing groups (–Cl, –F) substituents does not improve antioxidant activity. Our studies suggest that the presence of nitrile, 1,2,3-triazole ring and arylselenium moieties could contribute to the antioxidant activity of these compounds.
After these antioxidant assays, we returned our attention to verifying the toxicity of compounds 3a–e. To this end, δ-aminolevulinate dehydratase activity (δ-ALA-D) was determined. δ-ALA-D, a sulfhydryl- and zinc-containing enzyme, catalyzes the asymmetrical condensation of two molecules of δ-aminolevulinic acid (δ-ALA) to produce porphobilinogen (PBG) an intermediary in heme biosynthesis.24 δ-ALA-D is a sensitive enzyme inhibited in pro-oxidant situations and by oxidizing agents through oxidation of their sulfhydryl groups.25 Moreover, its activity is an important indicator of organochalcogen toxicity.26 The in vitro results obtained in this study (Tables 8–10) indicate that compounds did not inhibit δ-ALA-D activity, showing the lack of pro-oxidant activity of these compounds, once there is no oxidation of thiol groups to disulfide.
| Concentration (μM)/compound | 3a | 3b | 3c | 3d | 3e |
|---|---|---|---|---|---|
| 10 | 9.17 ± 0.71 | 8.92 ± 0.76 | 8.38 ± 0.75 | 10.55 ± 1.18 | 11.32 ± 1.43 |
| 50 | 9.05 ± 0.64 | 11.55 ± 0.48 | 10.06 ± 0.91 | 9.13 ± 0.28 | 10.74 ± 1.17 |
| 100 | 11.81 ± 0.57 | 11.09 ± 0.50 | 12.02 ± 0.96 | 11.07 ± 0.39 | 10.91 ± 0.13 |
| 500 | 10.61 ± 0.30 | 9.08 ± 0.61 | 9.13 ± 0.36 | 10.87 ± 0.40 | 11.58 ± 0.04 |
| Concentration (μM)/compound | 3a | 3b | 3c | 3d | 3e |
|---|---|---|---|---|---|
| 10 | 7.06 ± 0.54 | 8.09 ± 0.62 | 6.78 ± 0.26 | 8.65 ± 0.38 | 7.93 ± 0.17 |
| 50 | 6.68 ± 0.32 | 6.85 ± 0.55 | 7.02 ± 0.15 | 8.53 ± 0.37 | 7.96 ± 0.26 |
| 100 | 9.00 ± 0.59 | 7.84 ± 0.08 | 8.46 ± 0.24 | 10.28 ± 0.66 | 9.04 ± 0.50 |
| 500 | 8.65 ± 0.59 | 6.88 ± 0.52 | 10.32 ± 0.86 | 8.93 ± 0.39 | 9.56 ± 0.38 |
| Concentration (μM)/compound | 3a | 3b | 3c | 3d | 3e |
|---|---|---|---|---|---|
| 10 | 1.93 ± 0.12 | 2.57 ± 0.13 | 1.68 ± 0.02 | 2.01 ± 0.54 | 2.00 ± 0.10 |
| 50 | 2.44 ± 0.04 | 2.05 ± 0.12 | 1.73 ± 0.06 | 2.24 ± 0.24 | 2.11 ± 0.20 |
| 100 | 2.26 ± 0.14 | 1.72 ± 0.08 | 1.83 ± 0.13 | 2.21 ± 0.15 | 1.86 ± 0.19 |
| 500 | 2.86 ± 0.15 | 1.94 ± 0.12 | 1.85 ± 0.04 | 2.35 ± 0.13 | 1.91 ± 0.22 |
The selanyl-triazoyl carbonitriles (3) appear highly promising as intermediates for the preparation of more complex structures, such as tetrazoles. Tetrazoles have shown valuable properties in a wide range of applications, such as in material sciences,27 as propellants and explosives,28 catalysis,29 etc.30 Tetrazole derivatives are well known compounds with a high level of biological activity31 and are used in pharmaceuticals as carboxylic acids isosteres.32 An advantage of tetrazole derivatives over carboxylic acids is that they are resistant to various biological metabolic degradation pathways, conferring to the corresponding drug a longer bioavailability.32 Therefore, there is considerable interest in the development of efficient synthesis of highly functionalized and complex 1H-tetrazoles. In this regard, the most attractive way for their synthesis is the [2 + 3] cycloaddition involving nitriles and azides.33
A few years ago, our group developed the synthesis of selanyl substituted 1H-tetrazoles by treatment of arylselanyl–alkylnitriles with NaN3 in the presence of zinc bromide in aqueous solution.34 Attempts to extend the previously described conditions to the reaction between selanyl-triazoyl carbonitrile 3a and NaN3 to prepare the respective selanyl-triazoyl tetrazole 4a failed completely; even after 48 h under reflux (Scheme 3).
 | ||
| Scheme 3 First attempt to obtain selanyl-triazoyl tetrazole 4a. | ||
Nasrollahzadeh and co-workers described in 2002 the preparation of 5-substituted 1H-tetrazoles using FeCl3–SiO2 as a heterogeneous catalyst.35 Because of the complexity of our starting nitrile, we decide to adapt this procedure; however, we used Al2O3 as a solid support instead of SiO2. Thus, the reaction of selanyl-triazoyl carbonitrile 3a with NaN3 using FeCl3–Al2O3 as the catalyst in DMF at 120 °C for 48 hours under air atmosphere furnished the desired product 4a in 63% yield (Table 11, entry 1). Considering this a satisfactory result, we turned our attention to the evaluation of a range of other selanyl-triazoyl carbonitriles (3; Table 11).
|
|||
|---|---|---|---|
| Entry | Product (yield)b | Entry | Product (yield)b |
| a Reactions were performed with phenylselanyl-1H-1,2,3-triazole-4-carbonitriles 3a–f (0.3 mmol), NaN3 (0.45 mmol), Al2O3/FeCl3 as the catalyst (0.018 g), DMF as the solvent (1.5 mL) at 120 °C for 48 h under air atmosphere.b Yields are given for isolated products. | |||
| 1 |  |
4 |  |
| 2 |  |
5 |  |
| 3 |  |
6 |  |

The results shown in Table 11 demonstrate that selanyl-triazoyl carbonitriles 3b–f were efficiently converted to the respective tetrazole derivatives 4b–f. Therefore, selanyl-triazoyl carbonitriles containing EDG and EWG on the aromatic ring gave good yields of the desired selanyl-triazoyl tetrazoles 4b–f (Table 11, entries 2–6). All the spectral data (MS and NMR analysis) supported and confirmed the formation of the target compounds 4.
Conclusions
In conclusion, we demonstrated our results on the synthesis, antioxidant properties and synthetic application of phenylselanyl-1H-1,2,3-triazole-4-carbonitriles. This new class of compounds was synthesized in high yields by the reaction of azidophenyl phenylselenides with a range of α-keto nitriles in the presence of a catalytic amount of Et2NH. Five of the synthesized compounds were screened for their in vitro antioxidant activity and the results demonstrated that selanyl-triazoyl carbonitrile 3a is a better antioxidant than the other synthesized compounds. The selanyl-triazoyl carbonitriles were readily manipulated to yield the desired bifunctional hybrid selanyl-triazoyl tetrazoles in good yield by reaction with NaN3 in Al2O3/FeCl3 as a catalytic system. These synthetic methods proved to be efficient for the synthesis of new selenium-containing nitrogen heterocycles. Other bioassays are currently in progress to verify other possible activities of the selanyl-triazoyl carbonitriles derivatives as well as the mechanism involved.Experimental section
General remarks
Proton nuclear magnetic resonance spectra (1H NMR) were obtained at 400 MHz on a Bruker DPX-400 NMR spectrometer. Spectra were recorded in CDCl3. Chemical shifts are reported in ppm, with tetramethylsilane (TMS) used as the external reference. Data are reported as follows: chemical shift (δ), multiplicity, coupling constant (J) in Hertz and integrated intensity. Carbon-13 nuclear magnetic resonance spectra (13C NMR) were obtained at 100 MHz on a Bruker DPX-400 NMR spectrometer. Spectra were recorded in CDCl3. Chemical shifts are reported in ppm in reference to the solvent peak of CDCl3. Abbreviations to denote the multiplicity of a particular signal are s (singlet), d (doublet), t (triplet), dt (doublet of triplet) and m (multiplet). Mass spectra (MS) were measured on a Shimadzu GCMS-QP2010 mass spectrometer. High-resolution mass spectra (HRMS) were recorded on a Bruker Micro TOF-QII spectrometer 10416. Column chromatography was performed using a Merck Silica Gel (230–400 mesh). Thin layer chromatography (TLC) was performed using a 0.25 mm thick Merck Silica Gel GF254. For visualization, TLC plates were either placed under ultraviolet light or stained with iodine vapor or acidic vanillin.General procedure for the synthesis of phenylselanyl-1H-1,2,3-triazole-4-carbonitriles 3a–h
The appropriate α-keto nitrile (out of 2a–g, 0.3 mmol) was first added to a solution of the appropriate azidophenyl phenylselenide (out of 1a–b, 0.33 mmol) in DMSO (0.3 mL), followed by diethylamine (0.003 mmol) as the catalyst. The reaction mixture was stirred in an open vial for the time indicated in Table 1 and Scheme 2. After completion of the reaction, the crude product was purified by column chromatography on silica gel with a mixture of hexane/ethyl acetate (5:1) as the eluent to afford the desired product (3a–h). Spectral data for the products prepared are listed below.
General procedure for the synthesis of 5-aryl-1-(2-(phenylselanyl)phenyl)-1H-1,2,3-triazol-4-yl-1H-tetrazoles 4a–f
A mixture of the appropriate phenylselanyl-1H-1,2,3-triazole-4-carbonitrile (out of 3a–f, 0.3 mmol), sodium azide (0.5 mmol), Al2O3/FeCl3 (0.018 g) and DMF (1.5 mL) was taken in a round-bottomed flask and stirred at 120 °C for 48 h under air atmosphere. After this time, HCl (3 M, 2 mL) and ethyl acetate (10 mL) were added, and vigorous stirring was continued until no solid was present and the aqueous layer had a pH of 2. The resultant organic layer was separated and the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic layers were dried with MgSO4 and evaporated under reduced pressure. The resultant products were isolated in a chromatography column with hexane/ethyl acetate as the eluent and recrystallized if necessary to afford the desired product (4a–f). Spectral data for the products prepared are listed below.Acknowledgements
We are grateful to CAPES, CNPq (Grants 306430/2013-4, 400150/2014-0 and 447595/2014-8), FAPESP (15/17141-1 and 14/50249-8), FINEP and FAPERGS for financial support.Notes and references
- (a) A. R. Katritzky and A. F. Pozharskii, Handbook of Heterocyclic Chemistry, Pergamon, Oxford, 2000 Search PubMed; (b) T. Eicher and S. Hauptmann, The Chemistry of Heterocycles, Wiley, VCH, 2003 CrossRef; (c) L. Yu, M. Liu, F. Chen and Q. Xu, Org. Biomol. Chem., 2015, 13, 8379 RSC.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- (a) W. Dehaen and V. A. Bakulev, Chemistry of 1,2,3-triazoles, Springer International Publishing, New York, 1st edn, 2014 Search PubMed. For a recent set of reviews in this area, see the themed issues: (b) Chem. Soc. Rev., 2010, 39, 1221 Search PubMed; (c) Acc. Chem. Res., 2011, 44, 651 Search PubMed.
- (a) G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278 CrossRef CAS PubMed; (b) C. D. Hein, X. M. Liu and D. Wang, Pharm. Res., 2008, 25, 2216 CrossRef CAS PubMed; (c) J. Xie and C. T. Seto, Bioorg. Med. Chem., 2007, 15, 458 CrossRef CAS PubMed; (d) T. Lee, M. Cho, S. Y. Ko, H. J. Youn, D. J. Baek, W. J. Cho, C. Y. Kang and S. Kirn, J. Med. Chem., 2007, 50, 585 CrossRef CAS PubMed; (e) B. Parrish and T. Emrick, Bioconjugate Chem., 2007, 18, 263 CrossRef CAS PubMed; (f) N. Pokhodylo, O. Shyyka and V. Matiychuk, Med. Chem. Res., 2014, 23, 2426 CrossRef CAS.
- R. Huisgen, Angew. Chem., 1963, 75, 604 CrossRef CAS.
- For representative examples on copper or ruthenium catalysis see: (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef; (c) A. Krasinski, Z. Radic, R. Manetsch, J. Raushel, P. Taylor, K. B. Sharpless and H. C. Kolb, J. Am. Chem. Soc., 2005, 127, 6686 CrossRef CAS PubMed; (d) L. V. Lee, M. L. Mitchell, S. Huang, V. V. Fokin, K. B. Sharpless and C. Wong, J. Am. Chem. Soc., 2003, 125, 9588 CrossRef CAS PubMed; (e) J. E. Hein, J. P. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 1 CrossRef PubMed; (f) L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998 CrossRef CAS PubMed; (g) B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923 CrossRef CAS PubMed.
- (a) J. A. Johnson, J. M. Baskin, C. R. Bertozzi, J. T. Koberstein and N. J. Turro, Chem. Commun., 2008, 3064 RSC; (b) J. M. Baskin and C. R. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211 CrossRef CAS.
- (a) K. Jomova and M. Valko, Toxicology, 2011, 283, 65 CrossRef CAS PubMed; (b) J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. Carell, Org. Lett., 2006, 8, 3639 CrossRef CAS PubMed; (c) E. Lallana, E. Fernandez-Megia and R. Riguera, J. Am. Chem. Soc., 2009, 131, 5748 CrossRef CAS PubMed; (d) A. J. Link, M. K. S. Vink and D. A. Tirrell, J. Am. Chem. Soc., 2004, 126, 10598 CrossRef CAS PubMed; (e) L. M. Gaetke and C. K. Chow, Toxicology, 2003, 189, 147 CrossRef CAS PubMed; (f) D. C. Kennedy, C. S. McKay, M. C. B. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993 CrossRef CAS PubMed.
- For a feature article see: (a) C. G. S. Lima, A. Ali, S. S. van Berkel, B. Westermann and M. W. Paixão, Chem. Commun., 2015, 51, 10784 RSC; (b) S. S. V. Ramasastry, Angew. Chem., Int. Ed., 2014, 53, 2 CrossRef PubMed; (c) J. John, J. Thomas and W. Dehaen, Chem. Commun., 2015, 51, 10797 RSC.
- (a) N. Seus, L. C. C. Gonçalves, A. M. Deobald, L. Savegnago, D. Alves and M. W. Paixão, Tetrahedron, 2012, 68, 10456 CrossRef CAS; (b) N. Seus, B. Goldani, E. J. Lenardão, L. Savegnago, M. W. Paixão and D. Alves, Eur. J. Org. Chem., 2014, 1059 CrossRef CAS.
- (a) C. W. Nogueira and J. B. T. Rocha, Organoselenium and Organotellurium Compounds: Toxicology and Pharmacology, in Patai's Chemistry of Functional Groups, ed. Z. Rappoport, Wiley, Chichester, 2011 Search PubMed; (b) M. Iwaoka, Antioxidant Organoselenium Molecules, in Organoselenium Chemistry: Between Synthesis and Biochemistry, ed. C. Santi, Bentham Science, 2014, e-book, DOI:10.2174/97816080583891140101; (c) S. Santoro, J. B. Azeredo, V. Nascimento, L. Sancineto, A. L. Braga and C. Santi, RSC Adv., 2014, 4, 31521 RSC; (d) C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255 CrossRef CAS PubMed.
- (a) E. E. Alberto and A. L. Braga, Selenium and Tellurium Chemistry – From Small Molecules to Biomolecules and Materials, ed. W. J. Derek and L. Risto, Springer Verlag, Berlin, Heidelberg, 2011 Search PubMed; (b) T. Wirth, Organoselenium Chemistry: Synthesis and Reactions, Wiley-VCH, Weinheim, 2011 Search PubMed; (c) P. H. Menezes and G. Zeni, Vinyl Selenides, in Patai's Chemistry of Functional Groups, John Wiley & Sons, 2011 Search PubMed; (d) F. A. Devillanova, Handbook of Chalcogen Chemistry: New Perspectives in S, Se and Te, Royal Society of Chemistry, Cambridge, UK, 2006 Search PubMed; (e) G. Perin, E. J. Lenardão, R. G. Jacob and R. B. Panatieri, Chem. Rev., 2009, 109, 1277 CrossRef CAS PubMed; (f) A. A. Vieira, J. B. Azeredo, M. Godoi, C. Santi, E. N. da Silva Júnior and A. L. Braga, J. Org. Chem., 2015, 80, 2120 CrossRef CAS PubMed; (g) L. Yu, J. Ye, X. Zhang, Y. Ding and Q. Xu, Catal. Sci. Technol., 2015, 5, 4830 RSC; (h) S. Santoro, J. B. Azeredo, V. Nascimento, L. Sancineto, A. L. Braga and C. Santi, RSC Adv., 2014, 4, 31521 RSC; (i) L. Yu, Y. Wu, T. Chen, Y. Pan and Q. Xu, Org. Lett., 2013, 15, 144 CrossRef CAS PubMed.
- (a) M. Tiecco, L. Testaferri, C. Santi, C. Tomassini, F. Marini, L. Bagnoli and A. Temperini, Angew. Chem., Int. Ed., 2003, 42, 3131 CrossRef CAS PubMed; (b) T. G. Back, R. J. Bethell, M. Parvez, J. A. Taylor and D. Wehrli, J. Org. Chem., 1999, 64, 7426 CrossRef CAS; (c) H. A. Stefani, N. C. S. Silva, F. Manarin, D. S. Lüdtke, J. Zukerman-Schpector, L. S. Madureira and E. R. T. Tiekink, Tetrahedron Lett., 2012, 53, 1742 CrossRef CAS; (d) H. A. Stefani, D. M. Leal and F. Manarin, Tetrahedron Lett., 2012, 53, 6495 CrossRef CAS; (e) A. M. Deobald, L. R. S. Camargo, M. Hörner, O. E. D. Rodrigues, D. Alves and A. L. Braga, Synthesis, 2011, 2397 CAS; (f) N. Seus, M. T. Saraiva, E. E. Alberto, L. Savegnago and D. Alves, Tetrahedron, 2012, 68, 10419 CrossRef CAS; (g) M. T. Saraiva, N. Seus, D. Souza, O. E. D. Rodrigues, M. W. Paixão, R. G. Jacob, E. J. Lenardão, G. Perin and D. Alves, Synthesis, 2012, 44, 1997 CrossRef CAS.
- F. Donato, M. G. Gomes, A. T. R. Goes, N. Seus, D. Alves, C. R. Jesse and L. Savegnago, Life Sci., 2013, 93, 393 CrossRef CAS PubMed.
- B. Halliwell, Biochem. Soc. Trans., 2007, 35, 1147 CrossRef CAS PubMed.
- (a) L. S. Duvvuri, S. Katiyar, A. Kumar and W. Khan, Expert Opin. Drug Delivery, 2015, 13, 1 Search PubMed; (b) L. Marseglia, S. Manti, G. D'Angelo, A. Nicotera, E. Parisi, G. Di Rosa, E. Gitto and T. Arrigo, Int. J. Mol. Sci., 2014, 16, 378 CrossRef PubMed; (c) R. Thanan, S. Oikawa, Y. Hiraku, S. Ohnishi, N. Ma, S. Pinlaor, P. Yongvanit, S. Kawanishi and M. Murata, Int. J. Mol. Sci., 2014, 16, 193 CrossRef PubMed.
- (a) L. X. Yang, L. J. Zhang, K. X. Huang, L. X. Kun, X. Y. Wang, J. Stockigt and Y. Zhao, J. Enzyme Inhib. Med. Chem., 2009, 24, 425 CrossRef CAS PubMed; (b) P. C. Nobre, E. L. Borges, C. M. Silva, A. M. Casaril, D. M. Martinez, E. J. Lenardão, D. Alves, L. Savegnago and G. Perin, Bioorg. Med. Chem., 2014, 22, 6242 CrossRef CAS PubMed; (c) E. A. Wilhelm, C. F. Bortolatto, C. R. Jesse and C. Luchese, Biol. Trace Elem. Res., 2014, 162, 200 CrossRef CAS PubMed; (d) R. Chandramohan, L. Pari, A. Rathinam and B. A. Sheikh, Chem.-Biol. Interact., 2015, 229, 44 CrossRef CAS PubMed.
- R. Gozzelino, Curr. Alzheimer Res., 2015, 12, 174 Search PubMed.
- D. J. Piñero and J. R. Connor, Neuroscientist, 2000, 6, 435 CrossRef.
- (a) D. Barreca, E. Bellocco, C. Caristi, U. Leuzzi and G. Gattuso, Food Res. Int., 2011, 44, 2190 CrossRef CAS; (b) J. P. Vargas, L. M. Pinto, L. Savegnago and D. S. Lüdtke, J. Braz. Chem. Soc., 2015, 26, 810 CAS.
- J. Harvey, S. C. Hardy and M. L. J. Ashford, Br. J. Pharmacol., 1999, 126, 51 CrossRef CAS PubMed.
- R. E. Huie and S. Padmaja, Free Radical Res. Commun., 1993, 18, 195 CrossRef CAS PubMed.
- (a) M. P. Mattson, Exp. Gerontol., 2010, 44, 625 CrossRef PubMed; (b) B. S. Mandavilli and K. S. Rao, Biochem. Mol. Biol. Int., 1996, 40, 507 CAS; (c) S. Y. Yu, M. Zhang, J. Luo, L. Zhang, Y. Shao and G. Li, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2013, 45, 47 CrossRef CAS PubMed.
- E. K. Jaffe, J. Bioenerg. Biomembr., 1995, 27, 169 CrossRef CAS PubMed.
- P. M. Chagas, C. F. Bortolatto, E. A. Wilhelm and C. W. Nogueira, J. Appl. Toxicol., 2013, 33, 480 CrossRef CAS PubMed.
- C. W. Nogueira, V. C. Borges, G. Zeni and J. B. T. Rocha, Toxicology, 2003, 191, 169 CrossRef CAS PubMed.
- (a) B. C. Tappan, M. H. Huynh, M. A. Hiskey, D. E. Chavez, E. P. Luther, J. T. Mang and S. F. Son, J. Am. Chem. Soc., 2006, 128, 6589 CrossRef CAS PubMed; (b) M. B. Talawar, A. P. Agrawal, M. Anniyappan, D. S. Wani, M. K. Bansode and G. M. Gore, J. Hazard. Mater., 2006, 137, 1074 CrossRef CAS PubMed; (c) G. H. Tao, Y. Guo, Y. H. Joo, B. Twamley and J. M. Shreeve, J. Mater. Chem., 2008, 18, 5524 RSC; (d) G. I. Koldobskii and V. A. Ostrovskii, Usp. Khim., 1994, 63, 847 CrossRef CAS.
- (a) D. Moderhack, J. Prakt. Chem., 1988, 340, 687 CrossRef; (b) V. A. Ostrovskii, M. S. Pevzner, T. P. Kofmna, M. B. Shcherbinin and I. V. Tselinskii, Targets Heterocycl. Syst., 1999, 3, 467 CAS; (c) M. Hiskey, D. E. Chavez, D. L. Naud, S. F. Son, H. L. Berghout and C. A. Bome, Proc. Int. Pyrotech. Semin., 2000, 27, 3 Search PubMed.
- (a) A. J. A. Cobb, D. M. Shaw and S. V. Ley, Synlett, 2004, 558 CAS; (b) S. B. Zhou, Y. Q. Zhou, Y. L. Xing, N. X. Wang and L. H. Cao, Chirality, 2011, 23, 504 CrossRef CAS PubMed; (c) E. Barbayianni, P. Bouzi, V. Constantinou-Kokotou, V. Ragoussis and G. Kokotos, Heterocycles, 2009, 78, 1243 CrossRef CAS; (d) A. Odedra and P. H. Seeberger, Angew. Chem., Int. Ed., 2009, 48, 2699 CrossRef CAS PubMed; (e) H. A. Dabbagh, A. Najafi-Chermahini and S. Banibairami, Tetrahedron Lett., 2006, 47, 3929 CrossRef CAS.
- (a) R. P. Singh, R. D. Varma, D. T. Meshri and J. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45, 3584 CrossRef CAS PubMed; (b) Y. H. Joo and J. M. Shreeve, Angew. Chem., Int. Ed., 2009, 48, 564 CrossRef CAS PubMed; (c) Y. H. Joo and J. M. Shreeve, Angew. Chem., Int. Ed., 2010, 49, 7320 CrossRef CAS PubMed; (d) Y. H. Joo and J. M. Shreeve, Chem.–Eur. J., 2009, 15, 3198 CrossRef CAS PubMed; (e) Y. Guo, G. H. Tao, Z. Zeng, H. Gao, D. A. Parrish and J. M. Shreeve, Chem.–Eur. J., 2010, 16, 3753 CrossRef CAS PubMed; (f) D. E. Chavez and M. A. Hiskey, J. Energ. Mater., 1999, 17, 357 CrossRef CAS.
- (a) R. Siles, Y. Kawasaki, P. Ross and E. Freire, Bioorg. Med. Chem. Lett., 2011, 21, 2305 CrossRef PubMed; (b) B. J. Al-Hourani, S. K. Sharma, J. Y. Mane, J. Tuszynski, V. Baracos, T. Kniess, N. Suresh, J. Pietzsch and F. Wuest, Bioorg. Med. Chem. Lett., 2011, 21, 1823 CrossRef CAS PubMed; (c) E. S. Schaffert, G. Hofner and K. T. Wanner, Bioorg. Med. Chem., 2011, 19, 6492 CrossRef CAS PubMed; (d) R. S. Upadhayaya, S. Jain, N. Sinha, N. Kishore, R. Chandra and S. K. Arora, Eur. J. Med. Chem., 2004, 39, 579 CrossRef CAS PubMed; (e) C. N. S. S. Kumar, D. K. Parida, A. Santhoshi, A. K. Kota, B. Sridhar and V. J. Rao, Med. Chem. Commun., 2011, 2, 486 RSC.
- (a) H. Singh, A. S. Chawla, V. K. Kapoor, D. Paul and R. K. Malhotra, Prog. Med. Chem., 1980, 17, 151 CrossRef CAS PubMed; (b) R. J. Hert, Bioorg. Med. Chem., 2002, 10, 3379 CrossRef.
- J. Roh, K. Vávrová and A. Hrabálek, Eur. J. Org. Chem., 2012, 6101 CrossRef CAS.
- F. M. Libero, M. C. D. Xavier, F. N. Victoria, P. S. Nascente, L. Savegnago, G. Perin and D. Alves, Tetrahedron Lett., 2012, 53, 3091 CrossRef CAS.
- M. Nasrollahzadeh, Y. Bayat, D. Habibi and S. Moshaee, Tetrahedron Lett., 2009, 50, 4435 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra22445d |
| This journal is © The Royal Society of Chemistry 2016 |