
Platinum nanocatalysts on metal oxide based supports for low temperature fuel cell applications
N. R. Elezovic*a,
V. R. Radmilovicb and
N. V. Krstajicb
aInstitute for Multidisciplinary Research, University of Belgrade, Kneza Viseslava 1, Belgrade, Serbia. E-mail: nelezovic@tmf.bg.ac.rs
bFaculty of Technology and Metallurgy University of Belgrade, Karnegijeva 4, Belgrade, Serbia
First published on 6th January 2016
Abstract
In this manuscript a survey of the contemporary research related to platinum nanocatalysts on metal oxide based supports for low temperature fuel cell applications is presented. Different carbon based supports, used as state of the art materials, are listed and discussed, as well. Although carbon based materials possess many desirable properties, such as high surface area, high conductivity and relatively low cost and easy synthesis, the large scale commercialization is limited by instability under accelerated stability testing, simulating real fuel cell operating conditions. To overcome these disadvantages of carbon supports, different metal oxide based ones have been studied and promising results are referenced. The most often used oxide based supports for low temperature fuel cell applications are presented in this review. Suitable discussion and future research related remarks are given, as well.
1. Introduction
Polymer electrolyte membrane fuel cells (PEMFCs) are promising candidates for an environmentally friendly and efficient energy conversion, with many prospective practical applications in portable, stationary and transport devices. In spite of researchers' efforts during last few decades, many challenges in this field still remain unresolved. The main issues are still insufficient catalysts activity and stability, to decrease high costs, and relatively low life time of PEMFCs. Therefore, activity, stability and durability are major problems in this field and key points limiting the commercialization.1 It is well known that the whole cell performance is significantly influenced by the oxygen reduction reaction, owing to its slow kinetics and much higher overpotential, if compared to fast anodic hydrogen oxidation. Namely, the thermodynamic reversible potential for oxygen reaction on Pt is 1.23 V. Reversible hydrogen electrode, while the real open circuit potential value established vs. the same reference electrode is about 0.3 V lower. This large potential loss was attributed to the fact that the surface of Pt was covered with oxygen containing species from water (e.g. OH), or other adsorbed anions. Thus, the main source of energy losses in fuel cells is the oxygen reduction reaction.2 Platinum nanocatalysts on high surface area carbon based supports exhibit the best catalytic activity and stability for reactions taking place in fuel cell: oxygen reduction and hydrogen oxidation.2–6 Pt nanoparticles homogeneously distributed over carbon support (commercially named Vulcan and Ketjen black) are state of the art catalyst materials for both anode and cathode reactions. However, high cost and scarcity of platinum are the main problems to be solved before commercialization of these clean energy providers. Many efforts have been made to achieve sufficient low Pt loading (mgPt cm−2) to gain satisfactory power density: 0.9–1.2 gPt kW−1, for stationary, un-interrupted power supply, while less than 0.4 gPt kW−1 for large scale commercialization in automotive applications is required.7 Limited durability caused by degradation of the carbon support, usually used in commercially available Pt catalysts, increases total fuel cell costs and reduces its life time.8 One of the key requirements for this class of fuel cells application is that the fuel cell must be tolerant of frequent start–stop cycling. During startup and shutdown procedure high potential difference is created, causing carbon corrosion and oxygen evolution on the air cathode. This mechanism has been called “reverse-current decay mechanism”.9 Namely, carbon corrosion could cause permanent carbon loss, loss of the catalytic activity, even the whole catalyst degradation. Platinum further accelerates the carbon corrosion rate, leading to agglomeration of the catalyst particles and severe degradation of fuel cell cathode. The key contemporary research activities in this field are: (a) development and characterization of new interactive metal oxide based supports with improved durability for fuel cells application (molybdenum, titanium oxide, tin oxide based supports); (b) development and characterization of new Pt nanocatalysts on above mentioned supports, to improve activity, to achieve commercially acceptable power density, mentioned above.2. Carbon supported Pt nanocatalysts – state of the art materials for low temperature fuel cells application
The most used supports for low temperature fuel cells catalysts are carbon based. Namely, different carbon based materials were applied as platinum nanoparticles support owing to their properties: high surface area, relatively good chemical stability and well known synthesis procedures. A numerous papers were published, describing kinetics of oxygen reduction and hydrogen oxidation reactions at carbon supported platinum nanocatalysts in both acid and alkaline solutions.10–15 Pt nanoparticles dispersed on different types of high surface area and electrical conducting carbon based supports were studied: carbon powder, carbon nanotubes, fullerenes are shown to have high electrocatalyst activity. The Pt loading has been reduced significantly with improved Pt utilization.10–15 It was also clearly indicated that carbon support contributes itself to the kinetics of oxygen reduction in alkaline electrolyte, while in acid its contribution could be neglected.16 The effect of Pt particle size on the oxygen reduction electrocatalysis in both electrolytes was correlated with the predominant facets of the platinum crystallites.16 Tamizhmani et al.17 and Markovic et al.18 emphasized the crucial role of anion adsorption affects that could be cause of discrepancy and disagreement on the results and conclusions, related to specific and mass activities of the carbon supported catalysts in different electrolytes. The effect of the platinum particle size on the catalytic activity of Pt/Vulcan catalysts for oxygen reduction reaction has been studied and the loss of catalytic activity with the particle size decrease was demonstrated.19 Namely, the specific activity was three times lower for very small particles. The activity decrease seems to be related to a strong adsorption of oxygenated species on very small particles: on cyclic voltammograms recorded for various platinum particle sizes a shift of desorption peak of the oxygenated species was observed.19 Paulus et al. developed thin-film rotating disk electrode method and its application in a rotating ring disk configuration (RRDE) to the investigation of the oxygen reduction reaction on a supported catalyst powder (Pt:Vulcan XC 72 carbon).20 RRDE measurements allowed, for the first time, the direct determination of the fraction of peroxide production during oxygen reduction on carbon supported Pt catalysts.20 The comparison of the catalyst activity in H2SO4 and HClO4 solution revealed a significant effect of (bi)sulfate adsorption on the ORR activity. Namely, the mass-specific current densities in the non-specifically adsorbing electrolyte were significantly higher than in 0.5 M H2SO4 while the fraction of H2O2 formation was almost the same.20As the Nafion polymer is used for polymer electrolyte fuel cells it implies demand to explore its influence on catalytic activity of the Pt nanocatalysts. A. Ayad et al. investigated the kinetics of the reduction of oxygen on platinum covered by a Nafion® film in sulfuric in order to determine to what extent the polymer electrolyte modifies this reaction.21 The cyclic voltammogram obtained for platinum covered with a Nafion® film was similar to that for bare platinum. Namely, the essential difference was that the hydrogen adsorption/desorption peaks were weaker, which indicates that some active sites had been filled in by the Nafion®. The proportion of active sites blocked by Nafion® was estimated from 15% to 20%.22 It was also shown that the presence of Nafion® did not modify the kinetic parameters of oxygen reduction. The influence of Nafion was investigated by Z. Liu et al.23 Nafion stabilized Pt nanoparticles showed considerable activity for the oxygen reduction but very low activity for the methanol oxidation reaction, in comparison to a conventional unsupported Pt black electrocatalyst. This indicates that Nafion stabilizer has strong inhibiting effect on the methanol oxidation reaction. Thus, Nafion-stabilized Pt nanoparticles are potential methanol tolerant electrocatalysts for the O2 reduction reaction in DMFCs.23 Curnick et al.24 reported a facile method for the synthesis of stable and well-dispersed Nafion stabilized colloidal Pt nanoparticles with sizes in the range 3–10 nm. The activity for oxygen reduction was comparable with the commercial E-Tek catalyst. Combined with the near-100% utilisation measured at lower overall ionomer content for the Nafion1-Pt/C A catalysts, this implies that Pt nanoparticles synthesised with Nafion1 as a stabilizer can be ‘tuned’ to have simultaneous access to the reactant gas, the electron conducting carbon support and the proton conducting polymer electrolyte in the catalyst layer.24
Mayrhofer et al.25 scrutinized the thin-film rotating disc electrode (TF-RDE) method for testing the catalytic activity of high surface area catalysts, especially oxygen reduction reaction at carbon supported platinum nanocatalysts. It was referred that carbon supported high surface area Pt catalysts exhibited better activity in comparison to polycrystalline platinum. The most important conditions that have to be fulfilled for accurate estimation of high surface area catalysts activity were emphasized:25 (a) for a meaningful comparison of catalysts, normalized activities have to be determined rather than simply comparing polarization curves. The normalization can be based on the Pt loading (mass activity) or surface area (specific activity); (b) for the determination of the mass activity the complete utilization of the catalyst is mandatory. That is, the ECA has to be linear dependent on the loading. For the determination of the specific activity the active surface area has to be evaluated by applying a proper method that also considers the capacity of the support; (c) the diffusion limited current id of the ORR has to be within 10% of the theoretical value; (d) the current at the potential of interest (for the ORR usually in the region of 0.85–0.9 VRHE) has to be within 10% and 80% of the diffusion limited current, ideally below the half wave potential. Mayrhofer et al.25 also suggested importance of determination optimal Pt loading for the rotating disc measurements. Namely, due to low Pt loading RDE could not be completely and homogenously covered. On the contrary, too high Pt loading implies that mass transport characteristics of the RDE are no longer satisfied. This is illustrated in Fig. 1.
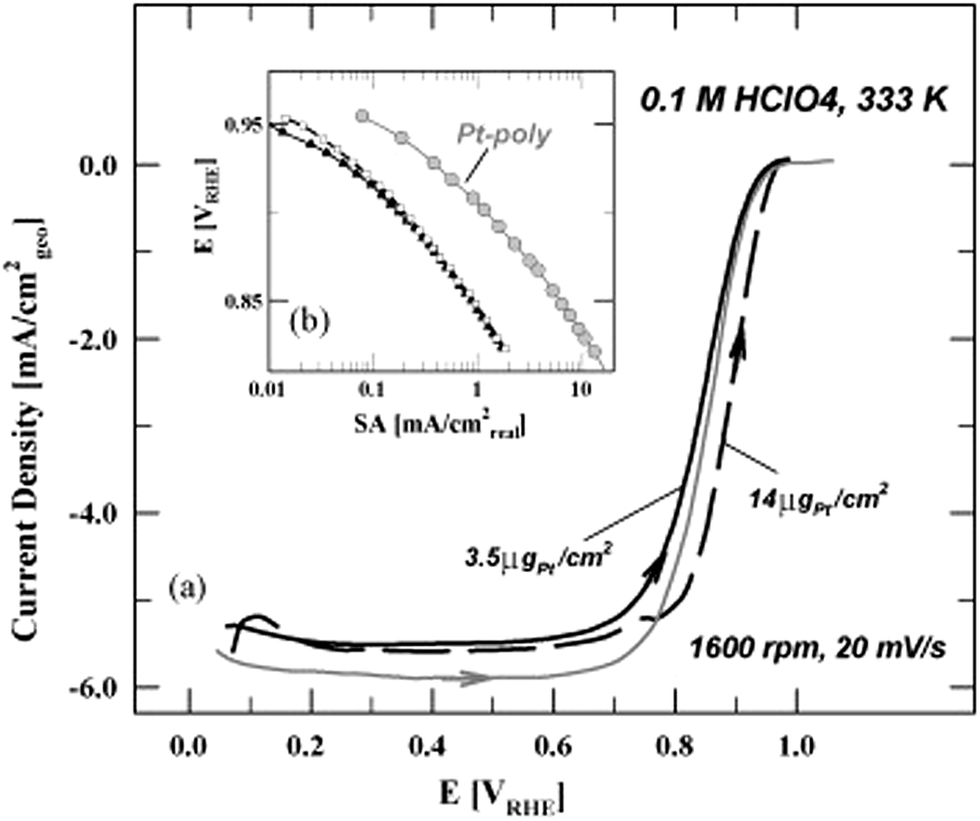 | ||
| Fig. 1 ORR-polarization (a) curves for Pt-poly and the 1 nm catalyst with two different Pt loadings in 0.1 M HClO4. The temperature is 333 K, sweep rate 20 mV s−1 and the rotation rate 1600 rpm. Only the anodic sweep is displayed for all samples. The estimated specific activities are presented in a Tafel-plot (b). Reprinted from the Electrochimica Acta in ref. 25, with permission of Elsevier. | ||
Yoshii et al.26 establishment of a facile Pt nanoparticle–SWCNT composite fabrication method that never requires a laborious pretreatment of SWCNTs or any chemical reagent was achieved by using Pt sputtered room temperature ionic liquid (RTIL). They found that RTIL can work as nanoglue for sticking Pt nanoparticles against SWCNTs. This approach enabled variations in the amount of Pt nanoparticle and Pt particle size supported on the Pt–SWCNT composite by changing the sputtering time and/or RTIL species in the Pt sputtering process Pt–SWCNT composite expressed a promising catalytic activity for electrochemical ORR.26 Speder et al.27 prepared Pt nanoparticles supported on high surface area carbon by a colloidal method. The electrocatalysts synthesized by this method have well-separated, size-controlled nanoparticles with tunable interparticle distance, and thus enable the examination of the particle proximity effect on the oxygen reduction reaction (ORR). The particle proximity effect implies that the activity of fuel cell catalysts depends on the distance between the catalyst particles and it was for the first time demonstrated for high surface area catalysts. Based on rotating disk electrode (RDE) experiments, it was shown that the kinetic current density of ORR depends on the distance between the neighboring nanoparticles, i.e. the ORR activity increases with decreasing interparticle distance.27 Singh et al.28 demonstrated a highly efficient approach for the effective dispersion and excellent decoration of Pt nanoparticles on both sides of carbon nanofibers, targeted to improve the Pt utilisation for methanol electrooxidation. This method has enabled the achievement of fine dispersion fine and excellent decoration of carbon nanofibers. The possibility of both side deposition improves the effective surface area as well as particle dispersion, increasing the efficiency and utilization of both nanofibers and Pt. This nanocatalysts could be very promising solutions to Pt dissolution/stability issues, indicating the antipoisoning strength and capability to be used in fuel cell applications.28 An ensemble of reduced functionalized graphene oxide, f-GO sheets and multiwalled carbon nanotubes (MWNTs), so called “few-layer graphene-MWNT sandwiches” (GCSs), were synthesized by a catalysis-assisted chemical vapor deposition (CCVD) method and explored as the electrocatalyst support material for oxygen reduction reaction (ORR) in a proton exchange membrane fuel cell (PEMFC).29 The Pt nanocatalyst deposited onto this support gave a maximum PEMFC performance of 495 mW cm−2 at 60 °C temperature. The improvement in the ORR activity was ascribed to the uniform dispersion of Pt nanoparticles with an optimal particle size (3.5 nm) over a well-organized conducting catalyst support.29 Pt-based nanoparticles on non-covalent functionalized carbon nanotubes were synthesized and characterized as effective electrocatalysts for proton exchange membrane fuel cells.30 It was referred that non-covalent functionalization or wrapping by polyelectrolytes introduced large number of active sites uniformly on the surface of outer walls of CNTs without generating oxygenated functional groups or breaking the C–C bonds of the graphene sheets of the CNTs. Pt-based nanoparticles can then be uniformly dispersed on CNTs via self-assembly principle with significantly reduced agglomerates.30 The well-defined geometry of outer wall surface of CNTs substantially promotes interconnectivity between Pt NPs, which leads to the significant increase in the grain boundaries between the NPs and thus active sites for fuel cell reactions.31 The mechanism of the influence non-covalent functionalized CNTs on the electrocatalytic activity of Pt-based NPs, particularly at the atomic scale, is still unknown. Some authors proved the synergistic interaction between Pt nanoparticles and CNT.32 Tian et al.33 investigated the structural and electronic properties of Pt nanoparticles on various nitrogen, (N)-doped graphene and their interaction with O by density functional theory (DFT) calculations. These results indicated that N-doped graphenes not only stabilized the Pt clusters but also enhanced their catalytic performance in the oxygen reduction reaction. Pd–Pt nanostructures were synthesized by a sequential reduction using an ethylene glycol reduction method and deposited on carbon nanofibers.34 The better performance in a single fuel cell was found with a Pd–Pt cathode than with a pure Pt commercial catalyst when both had the same loading of 0.5 mg of total metal per cm2. Therefore this catalyst, synthesized using the described method, is a promising alternative to pure Pt catalyst as a lower-cost catalyst for the cathode of a PEMFC. Jager introduced the novel non-conventional microporous–mesoporous carbide derived carbon powder, synthesized from molybdenum carbide (Mo2C) at 750 °C using the high-temperature chlorination method, and applied it as an electrocatalyst support for oxygen reduction.35 This catalyst showed noticeably higher activity expressed as the shift of half-wave potential (ΔE1/2 ∼ 50 mV) explained by the optimized hierarchical microporous/mesoporous electrode structure.
All above discussed literature results showed the advantages of platinum nanoparticle catalysts deposited on different types of carbon based supports. Although carbon based materials possess many desirable properties, such as high surface area, high conductivity and relatively low cost and easy synthesis, the large scale commercialization is limited by insufficient stability, especially at high positive potential values (1.4 V vs. RHE and higher). Degradation of the fuel cells catalyst during exposure to high anodic potentials has been already mentioned.8,9 Accelerated durability tests (ADTs) were carried out for a carbon-supported platinum catalyst using a rotating disk electrode at various rotation rates for various catalyst loadings by Nagai et al.36 The loss of the normalized ECSA increased with increasing the rotation rate and/or decreasing the catalyst loading. This analysis showed that the reciprocal of the normalized ECSA loss rate linearly increased with the increases in ω−1/2 and the catalyst loading, as it was presented in Fig. 2. These results suggested that ECSA loss in ADTs is significantly affected by Pt ion diffusion in the electrolyte.
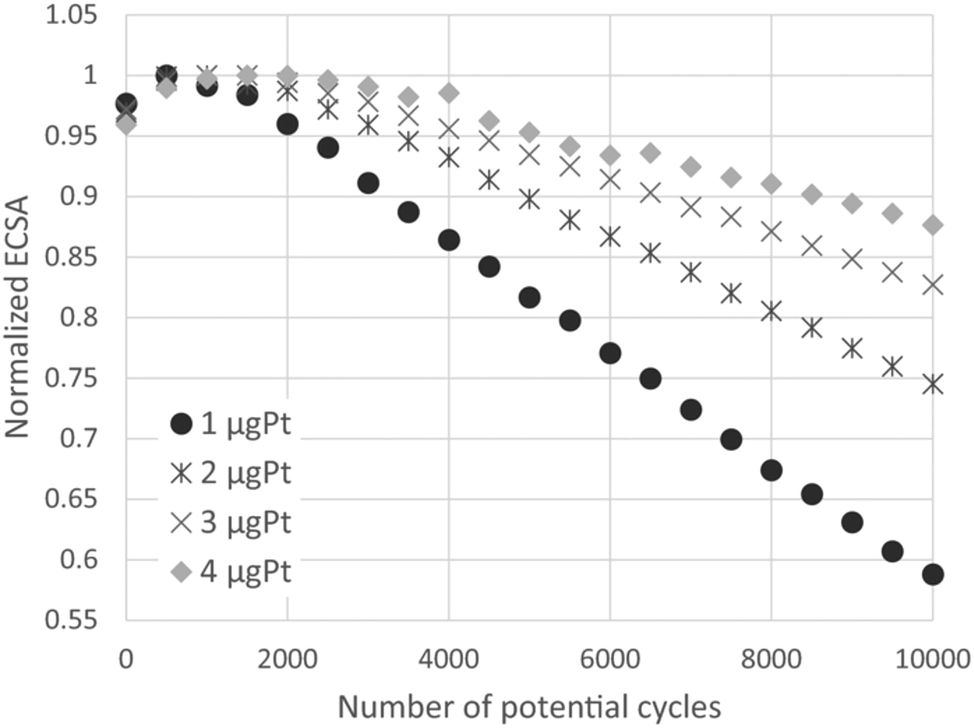 | ||
| Fig. 2 The normalized ECSA as a function of the potential cycles at 400 rpm for various catalysts loadings. The normalization is based on the maximum ECSA during the ADT. Reprinted with permission of The Electrochemical Society in ref. 36. | ||
An in situ electrochemical system for monitoring submonolayer dissolution of Pt under potential cycling was successfully established by Wang et al.37 The amount of dissolved Pt2+ was calculated using collector current (IC) and was confirmed by ex situ inductively coupled plasma mass spectrometry. The amount of Pt2+ and Pt4+ dissolved in different potential regions during potential cycling was quantified by IC for the first time. The mechanism of Pt dissolution under potential cycling in 0.5 M H2SO4 was proposed.37 Dhanushkodi et al.38 established a novel method to characterize durability of carbon supported platinum nanocatalysts in membrane electrode assembly. It was shown that during the accelerate stability testing (AST) the variations operating parameters (e.g., relative humidity, temperature and pressure), influenced the Pt dissolution rates and kinetic losses across the segmented cell. The Pt dissolution from the catalyst layer is shown to be non-homogenous during the AST via spatially resolved polarization resistance, kinetic polarization loss and the distance of the Pt band. Li et al.39 developed a one-dimensional model and studied platinum degradation and the subsequent electrochemical surface area (ECA) loss in the cathode catalyst layer (CL) of polymer electrolyte fuel cells (PEFCs). The model includes two mechanisms of Pt degradation: Ostwald ripening on carbon support and Pt dissolution–re-precipitation through the ionomer phase. Impact of H2|N2 or H2|air operation, operating temperature, and relative humidity on Pt degradation during voltage cycling was explored. It was shown that ECA loss is non-uniform across the cathode CL with a zone of exacerbated Pt degradation and hence much lower ECA found near the membrane. This non-uniform Pt degradation is caused by consumption of Pt ions by hydrogen crossover in both H2|N2 and H2|air systems. Improved performance of nitrogen-modified carbon supports for direct methanol fuel cell (DMFC) applications was referred by Olson et al.40 The nitrogen-ion implantation in high surface area carbon supports caused superior catalyst particle stability and performance as compared to industry standards. Specifically, results indicate a higher retention of metal catalyst surface area and electrochemical activity after accelerated electrochemical degradation testing.
3. Pt catalysts at metal oxide based supports for PEMFCs
In the previous section the contemporary research related to carbon based support was presented. It was clearly shown that carbon supported nanocatalysts suffer from serious problems caused by instability under accelerated stability testing, simulating real fuel cell operating conditions. This fact implies demand to introduce novel support materials, to overcome disadvantages of carbon based ones. For this purpose several types of metal oxide based supports have been studied and promising results were referred. The common required properties which make metal oxide suitable for platinum nanoparticles support are: high chemical stability in acid and alkaline electrolytes; high corrosion resistance-metal oxides should be electrochemically stable at fuel cells operating conditions; good electronic conductivity, to enable ultralow platinum loading; good proton conductivity; high surface area to enable high utilization of noble metal particles. The survey presenting the most often used oxide based supports for low temperature fuel cells application will be presented below.3.1. Molybdenum oxide supported Pt nanocatalysts
Investigations of molybdenum oxides have attracted remarkable researchers' interest for prospective fuel cells application. It is well known that molybdenum forms five Magneli phases oxide, with composition between MoO2 and MoO3. The presence of molybdenum oxide can significantly enhance activity and stability of noble metal catalysts by the strong metal support interaction, as well as by the formation of hydrogen molybdenum bronze.41 Chen et al.42 referred hydrogen molybdenum bronze formation and revealed hydrogen spillover on MoO3 in the presence of platinum. Elezovic et al.43,44 synthesized MoOx–PtC catalyst by the polyole method combined by MoOx post-deposition and characterized it for oxygen reduction and methanol oxidation reactions. Thermal treatment of this catalyst caused the loss in active surface area for about 26%, which was explained with oxide particle agglomeration in small three-dimensional islands on the neighboring Pt particles, decreasing the active surface area. The increase in catalytic activity for ORR on MoOx–Pt/C, in comparison with Pt/C catalyst, was explained by synergetic effects due to the formation of the interface between the platinum and oxide materials and by spillover due to the surface diffusion of oxygen reaction intermediates. The catalytic effect in oxidation of methanol is achieved only under potentiodynamic conditions, when poisoning species have no enough time to develop fully. Mixed valence state of molybdenum oxide was referred by Wang et al.,45 as well. They prepared Pt/MoOx catalysts by electrodeposition method and proposed promotion mechanism of MoOx for methanol oxidation reaction, causing removal of the adsorbed CO poisons at lower potentials. Justin et al. applied microwave assisted polyol method to incorporate MoO3 into Vulcan XC 72 particles.46 This catalyst exhibited high activity for methanol oxidation, as well as better stability than carbon supported one. The significant improvement in electrocatalytic activity and stability for the MOR was attributed to the strong metal-support interaction between Pt and MoO3, and the formation of hydrogen molybdenum bronze during the CV measurements, which reduces the CO poisoning of Pt catalysts. Papageorgopoulos et al.47 investigated the effect of the inclusion of Mo, Nb and Ta in Pt and PtRu carbon supported anode electrocatalysts on CO tolerance. Performed fuel cell tests demonstrated that while all the prepared catalysts exhibited enhanced performance compared to Pt/C, only the addition of a relatively small amount of Mo to PtRu results in a higher activity, in the presence of carbon monoxide. Ioroi et al.48 prepared carbon-supported Pt/Mo-oxide catalysts and examined the reformate tolerances of Pt/MoOx/C and conventional carbon supported anodes. Fuel cell performance was evaluated under various reformate compositions and operating conditions, and the CO concentrations at the anode outlet were analyzed simultaneously using on-line gas chromatography. Pt/MoOx showed better CO tolerance than PtRu with CO (80 ppm)/H2 mixtures, especially at higher fuel utilization conditions, which is mainly due to the higher catalytic activity of Pt/MoOx for the water-gas shift (WGS) reaction and electro-oxidation of CO. Ioroi et al.49 tested carbon-supported Pt/MoOx catalysts for the oxidation of CO contaminated H2, based on the fuel cell performance in PEFC single cell arrangements. Based on the XRD pattern and XPS measurements of the prepared catalysts, it was found that the deposited MoOx exists as an amorphous oxide phase. The MoOx phase shows a redox peak at around 0.45 V, which was revealed by the cyclic voltammogram of the Pt = MoOx = C in sulfuric acid solution. The PEFC performance of the cell with Pt = MoOx = C was improved under 100 ppm CO-contaminated H2 conditions compared to the Pt/C catalyst, and was almost comparable to the PtRu(1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)/C catalyst. Yan et al.50 synthesized the single molybdenum oxide (MoO2) crystals 5 nm in diameter on carbon, (denoted as C–MoO2) and tested structures, morphologies, chemical and electrocatalytic performances of the Pt nanoparticles supported on C–MoO2 (denoted as Pt/C–MoO2). High activity and stability for oxygen reduction was proven. A mass activity of 187.4 mA mg−1 Pt at 0.9 V is obtained for ORR, which is much higher than that on commercial Pt/C (TKK) electrocatalyst (98.4 mA mg−1 Pt). Furthermore, the electrochemical stability of Pt/C–MoO2 was higher than that of Pt/C. The origin of the improvement in catalytic activity was attributed to the synergistic or promotion effect of MoO2 on Pt.
:1)/C catalyst. Yan et al.50 synthesized the single molybdenum oxide (MoO2) crystals 5 nm in diameter on carbon, (denoted as C–MoO2) and tested structures, morphologies, chemical and electrocatalytic performances of the Pt nanoparticles supported on C–MoO2 (denoted as Pt/C–MoO2). High activity and stability for oxygen reduction was proven. A mass activity of 187.4 mA mg−1 Pt at 0.9 V is obtained for ORR, which is much higher than that on commercial Pt/C (TKK) electrocatalyst (98.4 mA mg−1 Pt). Furthermore, the electrochemical stability of Pt/C–MoO2 was higher than that of Pt/C. The origin of the improvement in catalytic activity was attributed to the synergistic or promotion effect of MoO2 on Pt.
3.2. Tungsten based supports for Pt nanocatalysts
Tungsten typically with oxygen forms stoichiometric tungsten trioxide, WO3, and lower non-stoichiometric oxides, WOx, where 2 < x < 3. Apart of WO3, only three stable tungsten oxides exist: β-oxide (W20O58), γ-oxide (W18O49), and δ-oxide (WO2).51 All other compounds are either metastable or consist of solid solutions of these three phases and WO3. WO3 is an n-type semiconductor with a reported band gap of about 2.6 to 2.8 eV.52 WO3 is insoluble in acid, while WO2 is soluble in acid and alkaline solution.53 In aqueous alkaline solutions WO3 forms tungstate ions, WO42−. Tungsten carbides (W2C and WC) are some of the hardest carbides, with melting point of 2770 °C for WC and 2780 °C for W2C. WC is an efficient electrical conductor, but W2C is less conductive. Tungsten carbide, as low cost alternative to bulk precious metal catalysts, is an excellent candidate support material for Pt monolayer, leading to good adhesion and utilizing the lowest possible Pt loading.54 Electrochemical stability of tungsten and tungsten carbide over wide pH and potential ranges have been studied and both materials showed promise as an anode, while operating within the region of passivation, and as a cathode, across the entire pH range.55Tungsten-based materials can play different roles in fuel cell systems. They are the only compounds which can be used as catalysts, co-catalysts, catalyst supports and electrolytes in different types of fuels cells. In particular, tungsten-based materials fulfill the requirements for their use as thermally stable carbon-alternative catalyst supports and Nafion®-alternative proton conducting electrolytes in fuel cells operating at intermediate temperature.56 Weigert et al. studied electrochemical behavior of tungsten carbide (WC) and platinum-modified WC as alternative DMFC electrocatalyst.57 Evidence of improved anode kinetics was observed by increasing the fuel concentration and operating temperature, particularly at 70 °C. It was evident that the most prominent limiting factor for the performance of membrane electrode assembly, prepared with this material, is the low surface area of the anode catalyst. Wang et al.58 prepared the high surface area (256 m2 g−1) tungsten carbide microspheres (TCMSs) by a simple hydrothermal method and studied performance of Pt electrocatalyst supported on the as-prepared TCMSs towards the oxygen reduction reaction (ORR). XRD results revealed presence both W2C and WC, while W2C exists as the main phase. It was found that the Pt particles are uniformly dispersed on the supports, while the corresponding average particle size was 3.7, 4.1 and 4.3 nm for Pt/C, Pt/CMSs and Pt/TCMSs, respectively. It was also found that in terms of ORR onset potential and mass activity, the Pt/TCMSs catalyst exhibits superior performance to that of Pt/CMSs and Pt/C. Catalytic activity improvement was attributed to higher electrochemical surface area (ESA), as well as to the synergistic effect between Pt and tungsten carbides. Hara et al.59 synthesized WC based support by the carburizing of tungsten nitrides (W2N) and tungsten sulfides (WS2) as precursors from different starting materials. Binary Pt–WC catalysts were prepared, with greatly improved performance by choosing a suitable WC and modifying the preparative method. As a result, it turned out that the mass activity of the Pt–WC catalyst system was superior to the current commercial Pt catalyst although only one-tenth the amount of Pt metal was used. It is thought that there is a synergistic effect between Pt and WC in terms of activity, leading to a drastic reduction in the use of Pt metal. Zhu et al.60 modified mesoporous carbon with tungsten carbides by the carbothermal hydrogen reduction of a layer of chemisorbed 1:12 phospho tungstic anions (PW12O403−). Depending on the temperature of carbothermal treatment, different tungsten species, i.e., W, W2C, WC, were formed on the carbon matrix. Uniform dispersion of Pt nanoparticles (1–6 nm) was achieved on the surfaces of both mesoporous carbon and tungsten carbide-modified mesoporous carbon. Pt nanoparticles supported on mesoporous carbons modified with tungsten carbide (Pt/WC–C) exhibit enhanced electrocatalytic activities relative to the control, in which mesoporous carbons without carbide modification were directly used as a support (Pt/C). In addition, both enhanced thermal stability and good electrochemical stability were observed for the Pt/WC–C electrocatalyst.60 The thermal and electrochemical stability of tungsten carbide (WC), with and without a catalyst dispersed on it, have been investigated to evaluate the potential suitability of the material as an oxidation-resistant catalyst support.61 Electrochemical testing was performed by applying oxidation cycles between +0.6 V and +1.8 V to the support-catalyst material combinations and monitoring the activity of the supported catalyst over 100 oxidation cycles. The electrochemical activity of the Pt/WC remained nearly constant over 100 accelerated oxidation cycles, while the activity of Pt/C was almost completely lost after only approximately 20 oxidation cycles. However, the initial activity of the Pt/WC supported catalyst is much lower than that of Pt/C at comparable volumetric catalyst loadings, indicating that higher surface area WC supports and better platinum dispersion techniques are still required in order to enhance the catalyst layer activity.61 Shengsheng et al. investigated oxidation resistance of tungsten carbide a catalysts support for proton exchange membrane fuel cells.62 The measurements described above showed that the Pt/WxCy catalyst possessed higher stability than the traditional Pt/C during the oxidation tolerance test owing to the excellent performance of the new support. Improvement of the surface area and rationalization of the pore distribution should be made to enhance the applicability of the tungsten carbide support. Mellinger et al.63 tested tungsten carbide and Pt-modified WC as CO-tolerant electrocatalysts as compared to pure Pt. The binding energies of CO, estimated from the desorption temperatures in the TPD measurements, are significantly lower on WC and Pt/WC surfaces than on pure Pt, indicating that the former two surfaces should be more CO-tolerant than Pt under typical PEMFC operating temperatures. The combination of low coverages of Pt with WC results in a synergistic effect for the oxidation of CO, leading to a higher activity than Pt and higher stability than WC. The promising properties of Pt/WC suggest the possibility of utilizing these materials either as CO-tolerant electrocatalysts or as purification catalysts to oxidize CO in the fuel feed stream. Weigert et al.64 examined the electrochemical stability of tungsten carbide (WC), Pt-modified WC, molybdenum carbide (Mo2C), and Pt-modified Mo2C using an in situ electrochemical half-cell in combination with X-ray photoelectron spectroscopy (XPS). At potentials higher than ∼0.8 V, WC oxidizes irreversibly into WxOy species as confirmed by the CV and XPS measurements. The deposition of submonolayer coverage of Pt on the WC surface increased the region of stability of WC, extending the onset of catalysts oxidation to ∼1.0 V (NHE). Therefore, Pt-modified WC can be a promising alternative electrocatalyst. Shen et al.65 introduced intermittent microwave heating (IMH) method for preparation of nanosized tungsten carbides. It produced WC with the average particle size of 21.4 nm at the procedure of 15 s-on and 15 s-off for 20 times, however, the particle size increased to 35.7 nm by CMH method for 5 min. The pure WC was obtained by post-treating the sample in NaOH solution, which gave the better performance as support. The nanosized WC was used as support for the Pt nanoparticles (Pt–WC/C (IMH)) for alcohol oxidation and oxygen reduction. It was proved that the Pt–WC/C (IMH) electrocatalysts gave the better performance than that prepared by CMH method (Pt–WC/C (CMH)) or Pt/C electrocatalysts in terms of the activity and CO-tolerance. Esposito and Chen66 discussed possibility to deposit monolayer platinum supported on tungsten carbides as low-cost electrocatalysts (ML Pt–WC). Density functional theory (DFT) calculations and experimental measurements indicate that the ML Pt–WC surface exhibits chemical and electronic properties that are very similar to bulk Pt for simple reactions such as the hydrogen evolution reaction, although deviation in behavior is observed for more complex reactions such as the electrooxidation of methanol. However, the challenges associated with the synthesis of such structures in a manner that can easily be scaled up in a cost-effective process are numerous. Ganesan et al.67 synthesized mesoporous WC with hexagonal crystal structure by a surfactant-assisted polymer method. A new electrocatalyst composed of a small amount of Pt supported on the mesoporous WC exhibited higher activity for electrooxidation of methanol than microporous Pt/WC or Pt/W2C as well as commercial Pt–Ru (1:1)/C catalysts. Xu et al.68 referred improving sulfur tolerance of noble metal catalysts by tungsten oxide-induced effects. WOx is adopted for the first time to modify such noble metal catalysts with an aim of acquiring excellent sulfur tolerance, due to its unique nature with hydrophilicity, redox couple in lower valence, as well as proton spillover effect. A series of WOx–Pt/C catalysts with various contents of tungsten oxide from 1 to 50 wt% were synthesized and compared to conventional Pt/C catalysts toward sulfur resistance using cyclic voltammetry (CV), as well as rotating ring disk electrode (RRDE) methods. Moreover, the higher catalytic activity of WOx–Pt/C toward oxygen reduction reaction (ORR) is revealed in comparison with Pt/C after both were poisoned by SOx where the electron transfer number of the former is closer to four-electron than that of the latter. The electronic interaction between Pt and WOx is evidently confirmed by X-ray photoelectron spectroscopy analysis and strongly suggested as the crucial factor for the ORR enhancement. Elezovic et al.69–71 synthesized and characterized platinum nanocatalysts on two tungsten based supports as the catalysts for oxygen reduction reaction in acid and alkaline solutions. Tungsten based support assigned WCctabr has been synthesized by polycondensation of resorcinol and formaldehyde in the presence of CTABr surfactant. Support assigned WCWO3 was synthesized from resorcinol/formaldehyde gel, using WO3 nanoparticles as starting material. Supporting materials have been characterized by BET (Brunauer, Emmett and Teller) technique and determined values of surface area were 80 m2 g−1 for WCctabr and 175 m2 g−1 for WCWO3. These catalysts exhibited better catalytic activity, expressed in terms of kinetic current density per real surface area at the constant potential and better stability, in comparison with Pt/C catalyst. Core–shell structure of WCctabr, containing W core and WC shell about 2–5 nm of thickness was confirmed by HRTEM technique (presented in Fig. 3).
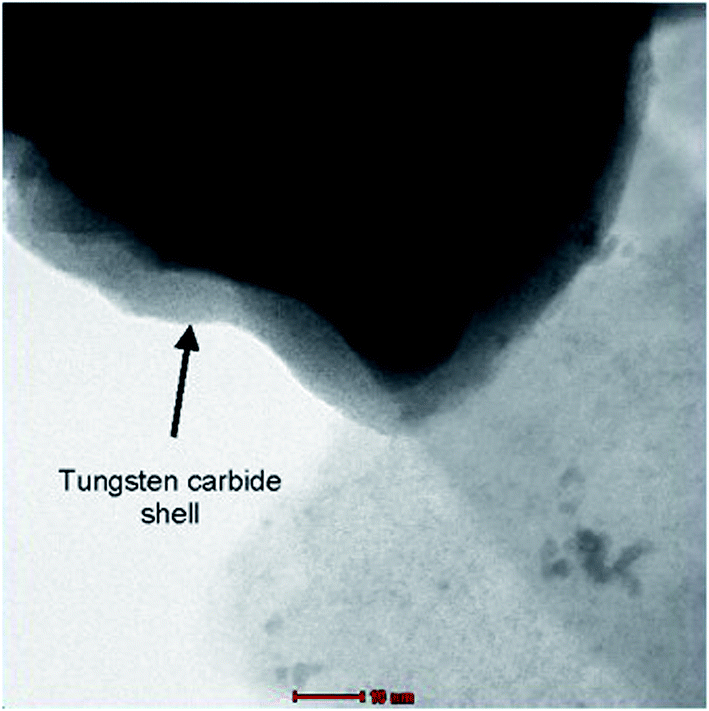 | ||
| Fig. 3 High resolution TEM micrograph of WCctabr support. Reprinted from Applied Catalysis B: Environmental in ref. 69, with permission of Elsevier. | ||
EDS analysis confirmed that shell was mixture of tungsten oxide and tungsten carbide. EELS have been performed as complementary technique to EDS spectroscopy, to confirm core–shell structure.
It is interesting to note that the existence of the Pt particles size lower than 2 nm, even clusters of Pt atoms were obtained (Fig. 4). This high resolution TEM image shows one-dimensional lattice fringes belonging to the (1 1 1) Pt plane (black square upper, Fig. 4b). Presence of tungsten particles oriented close to 100 zone axis is also indicated (black square lower, Fig. 4b).
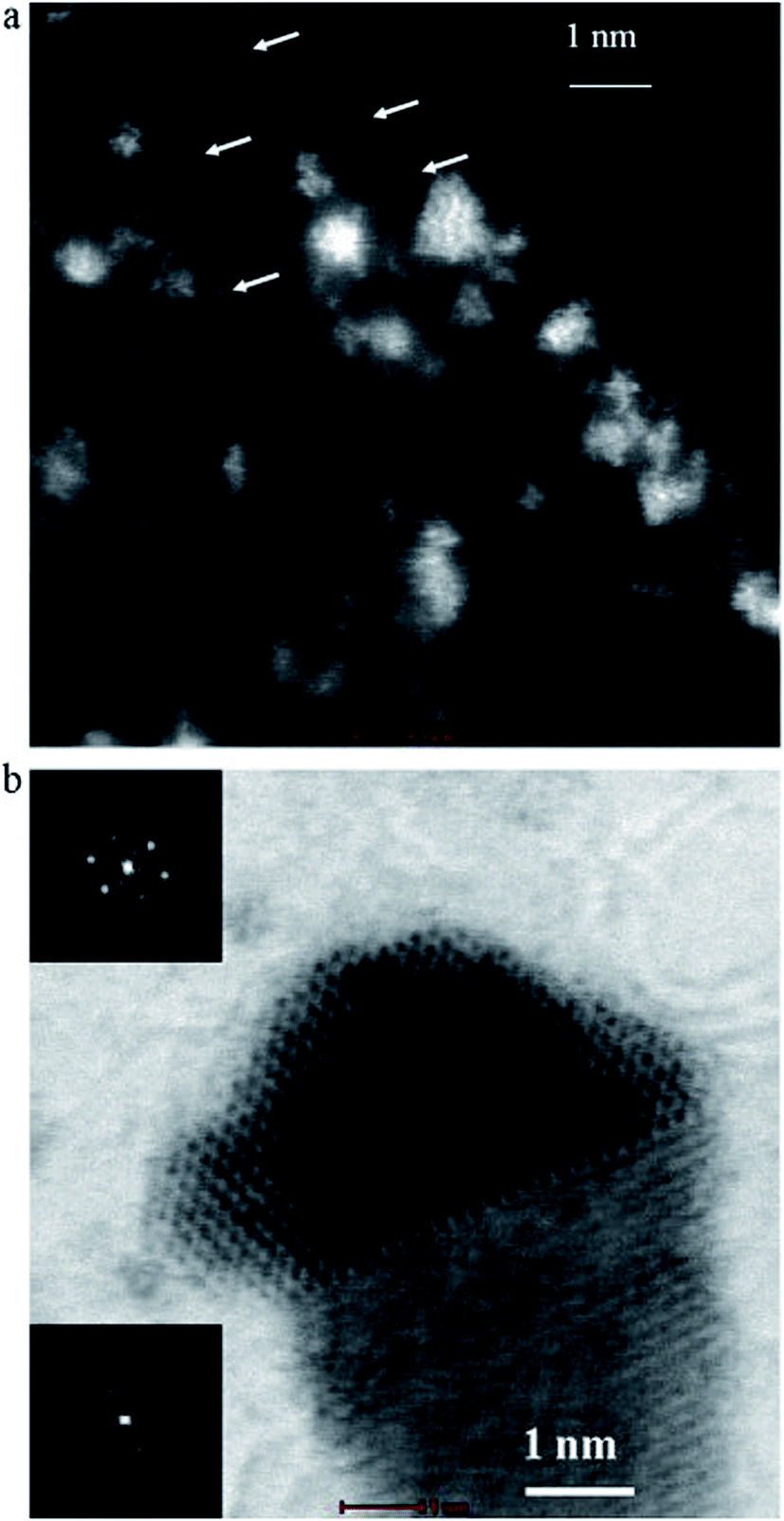 | ||
| Fig. 4 STEM images of the Pt/WC catalyst the smallest particles of Pt lower than 2 nm as clusters of Pt atoms. The arrows indicate individual Pt atoms, the indication Pt particle nucleated on WC support. Reprinted from Electrochimica Acta in ref. 70, with permission of Elsevier. | ||
Garcia et al.72 investigated the activity of Pt catalysts dispersed on tungsten carbide (WC) prepared with a high surface area carbon with two different WC/C ratios for the oxygen reduction reaction (ORR) in alkaline electrolyte. The PtWC-based catalysts show higher activity for the ORR compared to Pt/C, also involving a transfer of 4 electrons per oxygen molecule. CV and X-ray absorption near edge structure spectroscopy (XANES) results for the PtWC-based materials indicate weaker Pt–OHx interaction in these materials, resulting in a lower Pt-oxide coverage and explaining the increased rate of the ORR, as compared to Pt/C. Yan et al.73 presented an ion exchange route to produce WO3 nanobars as Pt electrocatalyst promoter for oxygen reduction reaction. Pt nanoparticles were supported on the C–WO3 composites (Pt/C–WO3) and used as electrocatalyst for ORR. The results showed that WO3 with moderate concentration could give the best promotion effect on Pt supported electrocatalyst. A typical Pt/C–WO3 (0.05) electrocatalyst gives the kinetic mass current of 174.6 mA mg−1 Pt, which is much higher than that of commercial Pt/C electrocatalyst (98.6 mA mg−1 Pt) for ORR at the same Pt loadings. Moreover, the electrochemical stability of Pt/C–WO3 was excellent. The higher catalytic activity and chemical stability may be due to the promotion effect of WO3 nanobars on Pt and the strong interaction force between WO3 and Pt respectively. Li et al.74 studied sub-stoichiometric tungsten oxide W18O49 as a support for a Pt catalyst. Pt/W18O49, achieved higher catalytic activity, antipoisoning properties and stability than the commercial Pt-black and Pt/C catalysts. XPS results revealed a strong metal-support interaction between Pt and the W18O49. In addition, two valence states of W (W5+ and W6+) were found to co-exist in W18O49, which may promote hydrogen spillover and oxygen buffering effects. These effects contributed to the improvement of the poisoning tolerance and the catalytic activity. Xiong et al.75 synthesized tungsten carbide support by the template of amorphous carbon microspheres (CMS), which is prepared by a hydrothermal synthesis process using glucose as the carbon precursor. The WC/CMS support is characterized by XRD, SEM, and BET. Pt nanoparticles were loaded on the WC/CMS support and Pt/WC/CMS catalyst was formed. The as-prepared Pt/WC/CMS showed higher electrocatalytic activity and anti-poisoning ability over Pt/C for the electrooxidation of ethanol. The rate constant of oxygen reduction reaction for Pt/WC/CMS is twice as high as that of Pt/C, indicating the synergetic effects between the Pt and the WC support. Furthermore, Pt/WC/CMS exhibits much higher stability for the ethanol electro-oxidation than Pt/C, which can be attributed to the high stability of WC support.
3.3. Titanium oxide based supports for Pt nanocatalysts
Titanium based oxides (commercially called Ebonex), with the integer formula TinO2n−1 (4 < n < 10), and between them the most pronounced their first two oxides, i.e. Ti4O7 and Ti5O9, have attracted a lot of attention as new catalyst supports.76,77 These oxides exhibit electrical conductivity and chemical stability at room temperature similar to these of carbon and graphite. The corrosion stability of these oxides in aqueous electrolytes is very high and they do not form hydrides in contact with hydrogen. All of that make them suitable as a support. Investigation of ORR at Ebonex supported Pt nanoparticles in 0.1 mol dm−3 NaOH solutions, revealed similar catalytic activity to polycrystalline Pt.77 High catalytic activity towards oxygen evolution at PtCo referred by Slavcheva et al.78 is related to the formation of surface oxides and electronic interactions between the metallic components of the catalyst and the supportive Ebonex. However, the problem with Ebonex powder was its very low specific surface area.78 Sasaki et al.79 demonstrated that niobium oxide nanoparticles could be adequate support for Pt, reducing at the same time the noble-metal contents of catalyst for oxygen reduction. Since niobium oxides have excellent chemical stability they could diminish the problems of substrate oxidation and corrosion degradation. It has also been reported that, by doping titania with pentavalent niobium ions, these ions get into the anatase titanium oxide crystalline structure preventing its phase transformation to rutile. This effect has been attributed to the extra valence of niobium ions, reducing oxygen vacancies in anatase phase and inhibiting the transformation to rutile. Park and Seul80 referred that NbTiO2 supported Pt catalyst showed an excellent catalytic activity for ORR in acid solution, mainly due to the good dispersion of Pt on NbTiO2. Elezovic et al.81,82 synthesized Nb doped TiO2 support by modified acid-catalyzed sol–gel procedure in non-aqueous medium. The catalytic activity and stability of Pt nanocatalysts synthesized onto this support were studied in both acid and alkaline solutions. This Nb–TiO2 (0.5% Nb) supported Pt nanocatalyst exhibited higher catalytic activity for ORR in acid solution, if compared with carbon supported Pt catalyst.81 Synthesized support surface area (91 m2 g−1) was much higher comparing with very low specific surface area of sub-stoichiometric titanium oxides as supporting material. Although the catalytic activities in alkaline solution were similar, the importance of new synthesized Nb doped TiO2 support, should be emphasized, as it was proved to be more stable than carbon based support.82 Xia et al. prepared high-stability Pt electrocatalysts using hierarchical carbon nanotubes, CNT@TiO2 structures composed of TiO2 nanosheets grafted on the CNT backbone as the support. The as-prepared Pt/CNT@TiO2 electrocatalysts manifest high electrocatalytic activity with greatly improved stability compared to conventional CNT or carbon black supported Pt electrocatalysts.83 Shi et al.84 achieved two times higher activity and three times higher stability in methanol oxidation reaction, a 0.12 V negative shift of the CO oxidation peak potential, and a 0.07 V positive shift of the oxygen reaction potential compared to Pt nanoparticles on pristine TiO2 support by tuning the electronic structure of the titanium oxide support of Pt nanoparticle catalysts. To explain the observed trends, an electronic structure model of Pt/TiOx electrocatalyst systems was introduced that shows an enhancement of the electrochemical activity of the Pt nanoparticles when the Fermi level of the support material is close to the Pt Fermi level and the redox potential of the reaction so that to facilitate electron transfer among the electrode, the Pt nanoparticles, and the electrolyte. Kumar and Ramani85 synthesized tantalum (Ta) modified TiO2 using a sol–gel procedure and evaluated for use as an electrocatalyst support. Pt supported on Ta0.3Ti0.7O2 was synthesized for further evaluation. The durability and catalytic activity of this material was measured and benchmarked against the state-of-the-art 46% Pt/C TKK electrocatalyst as well as 20% Pt/C catalyst prepared in-house. Ta0.3Ti0.7O2 exhibited an order of magnitude higher electrochemical stability than the benchmark carbon material when examined using an aggressive accelerated test protocol that simulated 10000 start-up and shut-down cycles (support stability test protocol: 1.0–1.5 V vs. RHE at a scan rate of 500 mV s−1 in a 0.1 MHClO4 electrolyte at 25 °C). 20% Pt/Ta0.3Ti0.7O2 also demonstrated greater electrochemical stability than both Pt/C catalysts, as estimated using the load cycling protocol, showing a 35% vs. a 44–47% loss in ECSA over 10000 load cycles (load cycling test protocol: 0.6–0.95 V vs. RHE with a dwell time of 3 s at either potentials in a 0.1 M HClO4 electrolyte at 25 °C). Siracusano et al.86 synthesized bare and Nb and Ta doped Ti-oxides by using various synthesis routes and investigated in terms of structure, morphology and electrochemical properties for application as catalyst supports in low temperature fuel cells. Non-carbonaceous supports prepared by colloidal procedures showed an excellent corrosion resistance, at least one order of magnitude better than Ketjenblack EC used as reference. The use of an oxide support appears promising to strongly improve the cathode catalyst stability especially under critical conditions that may occur in a fuel cell whereas suitable performance can be obtained by tailoring the catalyst preparation conditions. Zhang et al.87 demonstrated method for preparation of Pt/TiO2 nanotube arrays by H2 reduction. The effects of the reduction atmospheres and temperatures on Pt catalysts preparation were investigated. The well dispersion of Pt catalysts supported onto the TiO2 nanotube array exhibited favorable electrochemical performance and excellent durability. Tiido et al.88 prepared nano-sized Pt catalyst supported onto titanium dioxide functionalized graphene nanosheets (TiO2-FGSs) using the polyol method and utilized for a catalyst activity investigation toward the oxygen reduction reaction (ORR). The catalyst exhibited a relatively high electrocatalytic activity for the four-electron reduction of oxygen to water. Huang et al.89 dispersed Pt nanoparticles into mesoporous TiO2 thin films fabricated by a facile electro-chemical deposition method and tested it as electro-catalysts for oxygen reduction reaction. The XPS studies confirmed that the TiO2 support had a strong influence on the electronic structure of the platinum particles deposited, which in turn affected the composite's activity. The electrochemical deposition method can be a generally used approach to the preparation of metal NPs into oxide nanocrystalline mesoporous solid thin film, which has potential applications in fuel cell. Meenakshi et al.90 developed a new carbon supported HT–Pt–TiO2 composite catalyst comprising Pt and Ti in varying atomic ratio, namely 1:1, 2:1 and 3:1. ORR studies in the presence and absence of ethanol on HT–Pt–TiO2/C catalyst containing Pt and Ti in varying atomic ratio show that HT–Pt–TiO2 (2:1)/C exhibits higher ORR activity. DEFC employing HT–Pt–TiO2 (2:1)/C as cathode catalyst exhibits better performance than that with Pt/C. Therefore, HT–Pt–TiO2/C catalyst is a good cathode catalyst for ethanol tolerant-oxygen-reduction reaction in direct ethanol fuel cells. Savych et al.91 synthesized TiO2 nanofibres (TNF) and carbon nanofibres (CNF) by electrospinning/calcination. Electronic conductivity of TNF was improved by doping and specific thermal treatments. Pt/TNF showed superior electrochemical stability compared to Pt/CNF. Rigdon and Huang92 prepared carbon monoxide tolerant platinum electrocatalysts on niobium doped titania and carbon nanotube composite supports. Bifunctional reactivity of platinum can be improved by the addition of select metal oxides to the surface of multi-wall carbon nanotube supports. Specifically, titania was donor doped with niobium in construction of stable electrocatalysts used in anodes with the best tolerance to 100 ppm carbon monoxide during hydrogen oxidation. Ruiz-Camacho et al.93 synthesized Pt nanoparticles by chemical vapor deposition on carbon and TiO2–C substrates. Significant differences in the electrochemical results
and alcohols tolerance are observed in the samples prepared in comparison whit Pt/C commercial catalyst. The methanol tolerance of the catalysts synthesized was higher compared to the ethanol tolerance. The electrochemical activity of Pt/TiO2–C catalyst prepared with TiO2 rutile phase was not affected by the presence of alcohols in comparison with Pt/C samples. It is explained by the thermal treatment over Pt/TiO2–C during the synthesis process that produces a synergetic effect caused by the formation of the interface between the platinum and oxide materials where titanium oxide acts as a protecting agent of platinum nanoparticles. Jukk et al.94 prepared Pt nanoparticle-titanium dioxide/multi-walled carbon nanotube (Pt–TiO2/MWCNT) materials by combining chemical vapour deposition (CVD), atomic layer deposition (ALD) and magnetron sputtering techniques. It was proven that incorporating TiO2 into the structure of fuel cell catalyst supports can improve the electrocatalytic activity of Pt nanoparticles towards the ORR. The specific activity of Pt–TiO2/MWCNT catalysts for oxygen reduction was slightly higher than that of commercial Pt/C in acid media. The prepared composite materials showed also a substantial electrocatalytic activity for ORR in alkaline solution. Liu et al.95 studied the oxygen reduction reaction (ORR) activity of Pt90Au10/C, promoted by TiOx modification. After accelerated durability testing, the ORR activity of PtAu/C with 3 wt% TiOx is 2.7 times higher than that of the Pt/C, attributed to the suboxide formation and moderate surface Pt–Au ratio. Li et al.96 synthesized a new type of SnO2–TiO2 solid solution (TixSn1−xO2) support was prepared via a solvothermal method with substitution of Ti4þ by Sn4þ in the TiO2 lattice. Furthermore, the TixSn1−xO2 was combined with conventional carbon black (Vulcan XC-72) to prepare a hybrid support (TixSn1−xO2eC) for depositing Pt nanoparticles. The ratios of Sn vs. Ti in the solid-solution and TixSn1−xO2 vs. XC-72 were systematically optimized in terms of their performance as supports for methanol oxidation. The enhanced activity for Pt supported on Ti0.9Sn0.1O2–C is explained by high content of OH group on Ti0.9Sn0.1O2 along with the strengthened metal supports interactions. Both promote the oxidation of poisoning CO absorbed on Pt active sites. Odetola et al.97 referred enhanced activity and stability of Pt/TiO2/carbon fuel cell electrocatalyst prepared using a glucose modifier. Higher ORR activity achieved when TiO2 was prepared with glucose doped carbon. Catalyst prepared with TiO2 showed slower rates of Pt dissolution/agglomeration compared to Pt/C. Sui et al.98 reported a rapid method to synthesize titania nanotubes as the support for a Pt-based catalyst. The titania nanotubes can be obtained during 1200 s in an ethylene glycol system by the anodization method. Pt nanoparticles were successfully deposited on a mixture of carbon and as prepared TiO2 nanotubes by a microwave-assisted polyol process. These studies have shown that the Pt/C–TNTs–EG catalyst served as a highly efficient catalyst for the methanol electrooxidation reaction with better activity and durability than the commercial Pt/C. The enhanced performance could be attributed to the metal-support interactions and hydrogen spillover effect between the Pt nanoparticles and titania, the high corrosion resistance of titania, the good electronic conductivity due to the addition of carbon, and the good dispersion of the Pt nanoparticles on the titania nanotubes. The results reported herein suggest that titania nanotubes obtained by anodization have potential for applications in the future. Shintani et al.99 introduced novel strategy to mitigate cathode catalyst degradation during air/air startup cycling via the atmospheric resistive switching mechanism of a hydrogen anode with a platinum catalyst supported on tantalum-doped titanium dioxide. It was shown that cathode degradation during startup was reduced by use of Pt/Ti0.9Ta0.1O2−δ anode. The membrane electrode assembly (MEA) with the Pt/Ti0.9Ta0.1O2−δ anode showed higher performance than the MEA with the Pt/GCB anode after the air/air startup cycling (presented in Fig. 5).
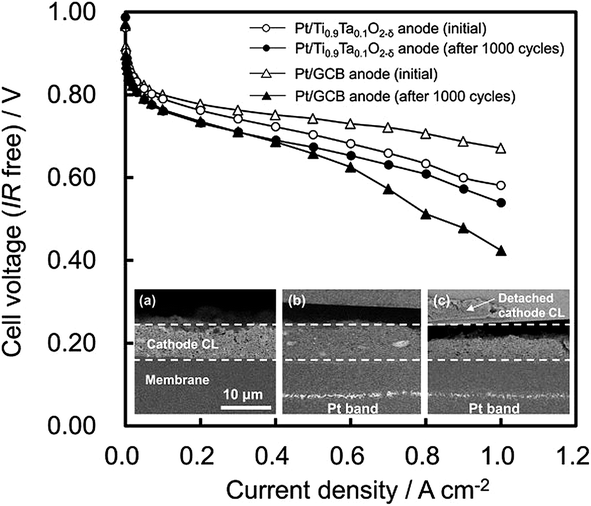 | ||
| Fig. 5 IR-free H2/air polarization curves of Pt/Ti0.9Ta0.1O2−δ anode cell (circles) and Pt/GCB anode cell (triangles) at 65 °C and 100% RH, initially (open symbols) and after the air/air startup cycling (solid symbols); H2 utilization 70%, oxygen utilization 40%, ambient pressure. The inset shows the cross-sectional SEM images of Pt/GCB cathode regions; for (a) the pristine MEA with Pt/GCB anode and the startup-tested MEAs with (b) Pt/Ti0.9Ta0.1O2−δ anode and (c) Pt/GCB anode. Reprinted from Journal of Power Sources in ref. 99, with permission of Elsevier. | ||
They concluded that the reverse current can be reduced by decreasing the ORR current generation on the Pt/Ta–TiO2 anode due to its high resistivity in air.
Elezovic et al.100,101 synthesized ruthenium doped TiO2 by hydrazine reduction method and apply it as a support for Pt nanoparticles. XRD analysis revealed mainly presence of anatase TiO2 phase and some peaks belonging to rutile TiO2. This new catalyst exhibited remarkable enhanced catalytic activity for oxygen reduction reaction in acid solution, compared to Pt/C. Increased catalytic activity of the Pt/Ru0.7Ti0.3O2 catalyst could be explained by effect of the support on the electronic structure of the catalyst due to the so called Strong Catalyst Support Interaction (SCSI). The hypo-d-electron transition metal oxides, such as TiO2, exhibit strong metal support interaction (SMSI) [100], which can improve the activity of the Pt catalyst towards the ORR. The modification of the electronic structure of Pt nanoparticles by interaction with the oxide interface results in a change in the adsorption characteristics of Pt on Ru–TiO2 support.
3.4. Tin oxide based supports for Pt nanocatalysts
SnO2, a broadband oxide semiconductor, with relatively high electronic conductivity, could be considered as a promising carbon-free catalyst support. The main advantages of tin oxide if compared to titanium based oxides are lower price of Sn than Ti and higher electrical conductivity. Santos et al.102 electrodeposited platinum micro particles on SnO2 thin films in order to verify the application of this system as a catalyst for the electrooxidation of methanol. The chronoamperometric results showed that the current values obtained for the electrooxidation of methanol were up to 10 times higher than the current values obtained with platinized platinum under the same conditions. Saha et al.103 prepared electrode by electrochemical deposition of platinum nanoparticles onto the surface of tin oxide SnO2 nanowires (NW) directly grown on the carbon fibers of a carbon paper. High electrocatalytic activities of the Pt/SnO2 NW/carbon paper composite electrode for both oxygen reduction reaction and methanol oxidation reaction have been achieved in comparison with standard Pt/C electrode. The higher electrocatalytic activities are attributed to the interaction of Pt nanoparticles with the SnO2 NW supports, as well as the 3D composite electrode structure. Lee et al.104 studied electrocatalytic activity and stability of Pt supported on Sb-doped SnO2 nanoparticles (Pt/ATO) for direct alcohol fuel cells. The activities of the Pt/ATO for both MOR and EOR were greater than those of the Pt/C as the amount of loaded Pt decreased. These enhanced activities can be attributed to better dispersion of Pt particles on the ATO support, as well as to the effects of SnO2 adjacent to Pt, such as the bifunctional effect and/or the electronic effect. Takasaki et al.105 used SnO2 as an alternative electrocatalyst support improving durability against voltage cycling up to a high potential, corresponding to the start-up and shut-down situation of polymer electrolyte fuel cell (PEFC) systems. Electrochemical surface area (ECSA) and oxygen reduction reaction (ORR) activity of Pt electrocatalysts as well as electrical conductivity of the electrocatalyst layers increase by doping of SnO2 with Nb or Sb. The durability tests with voltage cycles between 0.9 and 1.3 V versus reversible hydrogen electrode (RHE) potential have revealed that the Pt electrocatalyst supported on SnO2 (Pt/SnO2) withstands 60000 voltage cycles while maintaining its ECSA, which corresponds to a lifetime of more than 20 years with respect to the durability against voltage cycling (presented in Fig. 6).
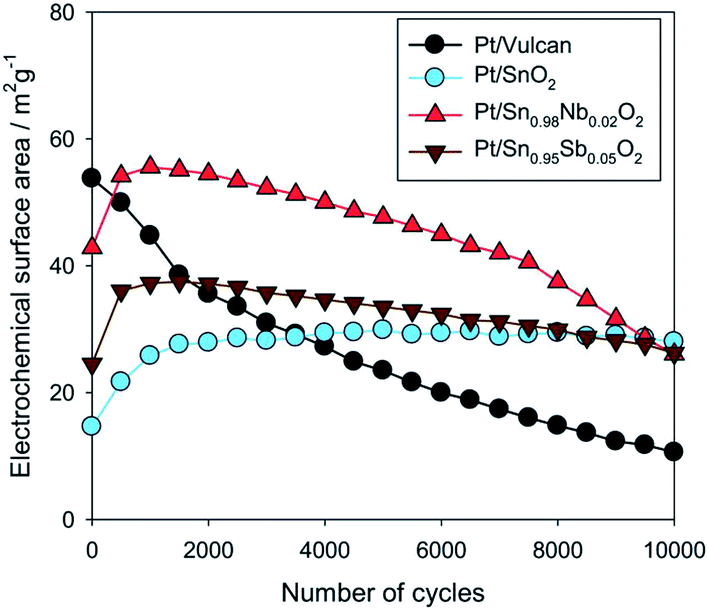 | ||
| Fig. 6 (Color online) ECSA vs. number of cycles in the potential range from 0.9 V to 1.3 V RHE, up to 10000 cycles. Reprinted from Journal of The Electrochemical Society in ref. 105, with permission of The Electrochemical Society. | ||
Nanostructure, electrochemical properties and durability of carbon-free electrocatalysts, Pt/SnO2, Pt/Nb–SnO2, Pt/Al–SnO2, were investigated by Takasaki et al.106 ECSAs of the electrocatalysts using the SnO2-based supports were lower than the ECSA of the conventional Pt/C. In comparison with Pt/SnO2, Pt/Sn0.95Nb0.05O2 exhibited slightly larger ECSA, whereas smaller ECSA was obtained for Pt/Sn0.95Al0.05O2. Kinetic current density of the electrocatalysts with the oxide supports was lower than that of Pt/C. Among the electrocatalysts examined, Pt/Sn0.95Nb0.05O2 exhibited the highest kinetic current value. In the durability test in the high potential range (0.6–1.3 VRHE) by CV, while the ECSA of Pt/C rapidly decreased down to nearly zero within 3000 times of voltage cycles, Pt/SnO2-based electrocatalysts exhibited considerably longer durability. Sasaki et al.107 prepared electrocatalysts with different oxide supports, Pt/SnO2, Pt/Nb–SnO2, Pt/Sb–SnO2, and Pt/Al–SnO2, as well as Pt/TiO2. It was demonstrated that electrocatalysts using alternative catalyst support materials exhibit comparable electrochemical performance to the conventional Pt/C electrocatalysts. Especially, the use of carbon-free oxide-supported electrocatalysts can be an ultimate solution to prevent the carbon corrosion problem. Kakinuma et al.108 synthesized a Pt catalyst supported on Sn0.96Sb0.04O2−δ with a random network structure for the cathode of the polymer electrolyte fuel cell (PEFC). The Sn0.96Sb0.04O2−δ support, synthesized by the flame combustion method, was in the form of nanometer-sized particles with a partially agglomerated structure similar to that of carbon black (CB) and with a high surface area, 125 m2 g−1. The ECA was 50.2 m2 g−1 (Pt) initially, and the values were maintained at a high value during the potential step cycle test (0.9–1.3 V). The oxygen reduction activity of the Pt/Sn0.96Sb0.04O2−δ catalyst exceeded that of Pt on carbon black. Fan et al.109 prepared hierarchical structure SnO2 with two-dimensional (2D) nanosheets building blocks as support material for Pt nanoparticles (NPs). The superior catalytic performance may arise from the unique multiscale structure and morphology of the SnO2 support, which process extraordinary promotional effect on Pt catalyst. Dou et al.110 synthesized tin oxide nanocluster (SnO2) with parallel nanorods via a hard template method and explored as the anode catalyst support for proton exchange membrane fuel cells (PEMFCs). Electrochemical measurements showed that Pt/SnO2 exhibited significantly enhanced electrochemical stability than Pt/C under high potential electro-oxidation and potential cycling. The Pt/SnO2 catalyst reserved most of its electrochemically active surface area (ECA) under 10 h potential hold at 1.6 V while its ECA degradation rate was one order of magnitude lower than Pt/C under potential cycling between 0.6 and 1.2 V. Elezovic et al.111,112 t synthesized two different tin oxide based supports, Sb–SnO2 and Ru–SnO2, by hydrazine reduction method. Pt catalysts on Sb and Ru doped SnO2 support exhibited catalytic activities comparable to Pt on commercial carbon based support. Stability tests were also performed. Determined small loss of electrochemical active surface area of the Pt catalyst on Sb doped tin oxide support, after repetitive cycling, indicated high stability and durability of this cathode for prospective fuel cells application. Hoque et al.113 made unique tin oxide-mesoporous carbon (SnO2–CMK-3) composites have been synthesized as platinum nanoparticle electrocatalyst supports for low temperature fuel cell applications. The improvements to the oxygen reduction reaction (ORR) kinetics were observed, with Pt/SnO2–CMK-3 providing a kinetic current density of 3.40 mA cm−2 at an electrode potential of 0.9 V vs. RHE. The improved performance of Pt/SnO2–CMK-3 for EOR and ORR was attributed to the beneficial impact of the support properties, along with potential interactions occurring between the support and catalyst particles. Zhang et al.114 referred a novel nanostructural Pt/SnO2/C catalyst synthesized by depositing Pt nanoparticles on the SnO2/C surface. Electrochemical measurements showed that specific activity towards oxygen reduction reaction (ORR) of Pt/SnO2/C catalysts was 2.3 times as high as that of conventional Pt/C catalysts. Accelerated degradation test (ADT) indicated that electrochemical stability of Pt/SnO2/C was twice as high as that of conventional Pt/C catalysts. The improved performance can be attributed to the presence of Pt/SnO2/C triple junction nanostructures, in which Pt nanoparticles were thermodynamically favored to deposit at the SnO2/C junctions, and then be anchored simultaneously by both SnO2 and C. Tsukatsune et al.115 showed that using tin oxide (SnO2) and niobium-doped tin oxide (Nb–SnO2) as alternative electrocatalyst support materials can effectively solve the issue of carbon corrosion in polymer electrolyte fuel cell (PEFC) cathodes. The dependence of ECSA and ORR activity on Pt particle size, support surface area, Pt loading and residual Cl− ion contamination was examined. The ECSA of Pt/SnO2 and Pt/Nb–SnO2 electrocatalysts was comparable to Pt/Vulcan after optimization of the normalized support surface area. However, in this study, since the specific surface area of supports was not large enough, an increase in ECSA derived by decreasing Pt particles size resulted in a decrease in specific activity. Nb-doping of the SnO2 support also increased the specific activity, due to improved electronic conductivity. Consequently, oxide-supported PEFC electrocatalysts with high ORR activity require oxide supports with; (i) large specific surface area, (ii) high electronic conductivity, and (iii) negligible impurities. Dou et al.116 prepared Sb-doped SnO2 (ATO) nanoparticles and used as highly stable catalyst support for proton exchange membrane fuel cells. The obtained results demonstrated significantly improved durability if compared to Pt/C. After 800 cycles between 0.6 and 1.2 V vs. NHE, the ORR mass activity of Pt/ATO dropped by 35.1%, which was much lower than that of Pt/C (78.4%). The enhanced electrochemical stability of Pt/ATO is attributed to the high stability of ATO support and the strong interaction between Pt and ATO.
4. Conclusion
Generally, the electrocatalytic activity of supported Pt and Pt–M alloy catalysts is referred as mass activity (MA) or specific activity (SA), i.e. it is normalized with respect to the mass (MA) or the surface area (SA) of the catalyst in the electrode. Regarding the catalysts supported on metal oxide materials, their mass activity was generally lower than that of the same catalysts supported on carbon, due to the lower surface area of the oxide substrates.117 Thus, to increase the mass activity of supported catalysts, research goals have to be addressed to the achievement of support with high surface area. Having in mind all above stated results, it could be concluded that some problems referred earlier,117 were resolved. Namely, the high surface area, one of the most important demands was achieved.81,108,111,116 Therefore, tin oxide and titanium oxide based supports were synthesized with sufficiently high surface area. High stability of these materials under accelerated stability tests is also achieved.85,105,106,114,115 The main remark related to prospective commercial application is that there is still small number of investigations and testing the Pt catalysts on metal oxide supports in membrane electrode assembly form. The most of the literature results on this topic is performed as single electrode (single electrochemical reaction) testing. The other important issue is related to proper determination of the catalytic activities for oxygen reduction, as the whole performance of fuel cell is much more influenced by its slow kinetics and high overpotential. To ensure that results are comparable to other literature values one should pay attention on the following: (a) the proper rotating disc electrode measurements – the optimal catalyst loading applied; (b) proper correction for IR drop ohmic resistance, especially in low concentration electrolytes, such as commonly used 0.1 mol dm−3 HClO4; (c) proper formation of the thin film catalyst from the ink, to ensure reproducibility.Acknowledgements
This work was financially supported by Ministry of Education, Science and Technological Development, Republic of Serbia, under contract number 172054. Electron microscopy characterization was performed at the National Center for Electron Microscopy, Lawrence Berkeley National Laboratory, which is supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract no. DE-AC02-05CH11231.References
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 CAS.
- U. A. Paulus, T. J. Schmidt, H. A. Gaisteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134–145 CrossRef CAS.
- F. Maillard, M. Martin, F. Gloaguen and J. M. Leger, Electrochim. Acta, 2002, 47, 3431–3440 CrossRef CAS.
- R. Benitez, A. M. Chaparro and L. Daza, J. Power Sources, 2005, 151, 2–10 CrossRef CAS.
- Q. Huang, H. Yang, Y. Tang, T. Lu and D. L. Akins, Electrochem. Commun., 2006, 8, 1220–1224 CrossRef CAS.
- Z. Liu, Z. Q. Tian and S. P. Jiang, Electrochim. Acta, 2006, 52, 1213–1220 CrossRef CAS.
- H. A. Gasteiger, J. E. Panels and S. G. Yan, J. Power Sources, 2004, 127, 162–171 CrossRef CAS.
- T. Toda, H. Igarashi, H. Uchida and M. Watanabe, J. Electrochem. Soc., 1999, 146, 3750–3756 CrossRef CAS.
- C. A. Reiser, L. Bregoli, T. W. Patterson, S. Y. Jung, J. D. Yang, M. L. Perry and T. D. Jarvi, Electrochem. Solid-State Lett., 2005, 8, A273–A276 CrossRef CAS.
- A. Pozio, R. F. Silva, M. de Francesco, F. Cardellini and L. Giorgi, Electrochim. Acta, 2002, 48, 255–262 CrossRef CAS.
- P. Ma, T. Haolin, M. Shichun and Y. Runzhang, J. Mater. Res., 2004, 19, 2279–2284 CrossRef.
- G. Girishkumar, K. Vinodgopal and P. V. Kamat, J. Phys. Chem. B, 2004, 108, 19960–19966 CrossRef CAS.
- Z. Q. Tian, S. P. Jiang, Y. M. Liang and P. K. Shen, J. Phys. Chem. B, 2006, 110, 5343–5350 CrossRef CAS PubMed.
- Z. Liu, X. Lin, J. Y. Lee, W. Zhang, M. Han and L. M. Gan, Langmuir, 2002, 18, 4054–4060 CrossRef CAS.
- K. Vinodgopal, M. Haria, D. Meisel and P. Kamat, Nano Lett., 2004, 4, 415–418 CrossRef CAS.
- J. Perez, E. R. Gonzalez and E. A. Ticianelli, Electrochim. Acta, 1998, 44, 1329–1339 CrossRef CAS.
- G. Tamizhmani, J. P. Dodelet and D. Guay, J. Electrochem. Soc., 1996, 143, 18–23 CrossRef CAS.
- N. M. Markovic, H. Gasteiger and P. N. Ross, J. Electrochem. Soc., 1997, 144, 1591–1597 CrossRef CAS.
- L. Genies, R. Faure and R. Durand, Electrochim. Acta, 1998, 44, 1317–1327 CrossRef CAS.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134–145 CrossRef CAS.
- A. Ayad, Y. Naimi, J. Bouet and J. F. Fauvarque, J. Power Sources, 2004, 130, 50–55 CrossRef CAS.
- S. K. Zecevic, J. S. Wainright, M. H. Litt, S. Y. Gojkovic and R. F. Savinell, J. Electrochem. Soc., 1997, 144, 2973–2982 CrossRef CAS.
- Z. Liu, Z. Q. Tian and S. P. Jiang, Electrochim. Acta, 2006, 52, 1213–1220 CrossRef CAS.
- O. J. Curnick, B. G. Pollet and P. M. Mendes, RSC Adv., 2012, 2, 8368–8374 RSC.
- K. J. J. Mayrhofer, D. Strmcnik, B. B. Blizanac, V. Stamenkovic, M. Arenz and N. M. Markovic, Electrochim. Acta, 2008, 53, 3181–3188 CrossRef CAS.
- K. Yoshii, T. Tsuda, T. Arimura, A. Imanishi, T. Torimotoe and S. Kuwabata, RSC Adv., 2012, 2, 8262–8264 RSC.
- J. Speder, L. Altmann, M. Baumer, J. J. K. Kirkensgaard, K. Mortensenc and M. Arenz, RSC Adv., 2014, 4, 14971–14978 RSC.
- B. Singh and E. Dempsey, RSC Adv., 2013, 3, 2279–2287 RSC.
- M. Sahoo, B. P. Vinayan and S. Ramaprabhu, RSC Adv., 2014, 4, 26140–26148 RSC.
- W. Yuan, S. Lu, Y. Xiang and S. P. Jiang, RSC Adv., 2014, 4, 46265–46284 RSC.
- S. Y. Wang, S. P. Jiang, T. J. White, J. Guo and X. Wang, J. Phys. Chem. C, 2009, 113, 18935–18945 CAS.
- J. G. Zhou, X. T. Zhou, X. H. Sun, R. Y. Li, M. Murphy, Z. F. Ding, X. L. Sun and T. K. Sham, Chem. Phys. Lett., 2007, 437, 229–232 CrossRef CAS.
- Y. Tian, Y. Liu, J. X. Zhao and Y. Ding, RSC Adv., 2015, 5, 34070–34077 RSC.
- A. Godinez-Garcia and D. F. Gervasio, RSC Adv., 2014, 4, 42009–42013 RSC.
- R. Jager, E. Hark, P. E. Kasatkin and E. Lust, J. Electrochem. Soc., 2014, 161, F861–F867 CrossRef.
- T. Nagai, H. Murata and Y. Morimoto, J. Electrochem. Soc., 2014, 161, F789–F794 CrossRef CAS.
- Z. Wang, E. Tada and A. Nishikata, J. Electrochem. Soc., 2014, 161, F380–F385 CrossRef CAS.
- S. R. Dhanushkodi, M. Schwager, D. Todd and W. Merida, J. Electrochem. Soc., 2014, 161, F1315–F1322 CrossRef CAS.
- Y. Li, K. Moriyama, W. Gu, S. Arisetty and C. Y. Wang, J. Electrochem. Soc., 2015, 162, F834–F842 CrossRef CAS.
- T. S. Olson, A. A. Dameron, K. Wood, S. Pylpenko, K. E. Hurst, S. Christensen, J. B. Bult, D. S. Ginley, R. O'Hayre, H. Dinh and T. Gennett, J. Electrochem. Soc., 2013, 160, F389–F394 CrossRef CAS.
- Z. Zhang, J. Liu, J. Gu, J. Su and L. Cheng, Energy Environ. Sci., 2014, 7, 2535–2558 CAS.
- L. Chen, A. C. Cooper, G. P. Pez and H. Cheng, J. Phys. Chem. C, 2008, 112, 1755–1758 CAS.
- N. R. Elezovic, B. M. Babic, V. R. Radmilovic, L. M. Vracar and N. V. Krstajic, Electrochim. Acta, 2009, 54, 2404–2409 CrossRef CAS.
- N. R. Elezović, B. M. Babić, V. R. Radmilović, S. Gojković, N. V. Krstajić and L. M. Vračar, J. Power Sources, 2008, 175, 250–255 CrossRef.
- Y. Wang, E. R. Fachini, G. Cruz, Y. Zhu, Y. Ishikawa, J. A. Colucci and C. R. Cabrera, J. Electrochem. Soc., 2001, 148, C222–C226 CrossRef CAS.
- P. Justin and G. Ranga Rao, Int. J. Hydrogen Energy, 2011, 36, 5875–5884 CrossRef CAS.
- D. C. Papageorgopoulos, M. Keijzer and F. A. de Bruijn, Electrochim. Acta, 2012, 48, 197–204 CrossRef.
- T. Ioroi, T. Akita, S. Yamazaki, Z. Siroma, N. Fujiwara and K. Yasuda, Electrochim. Acta, 2006, 52, 491–498 CrossRef CAS.
- T. Ioroi, N. Fujiwara, Z. Siroma, K. Yasuda and Y. Miyazaki, Electrochem. Commun., 2002, 4, 442–446 CrossRef CAS.
- Z. Yan, J. Xie, J. Jing, M. Zhang, W. Wei and S. Yin, Int. J. Hydrogen Energy, 2002, 37, 15948–15955 CrossRef.
- B. S. Hobbs and A. C. C. Tseung, J. Electrochem. Soc., 1975, 122, 1174–1177 CrossRef CAS.
- M. A. Butler, J. Appl. Phys., 1977, 48, 1914–1920 CrossRef CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, Pergamon, London, 1966, p. 280 Search PubMed.
- D. V. Esposito and J. G. Chen, Energy Environ. Sci., 2011, 4, 3900–3912 CAS.
- M. C. Weidman, D. V. Esposito, I. J. Hsu and J. G. Chen, J. Electrochem. Soc., 2010, 57, F179–F188 CrossRef.
- E. Antolini and E. R. Gonzalez, Appl. Catal., B, 2010, 96, 245–266 CrossRef CAS.
- E. C. Weigert, S. Arisetty, S. G. Advani, A. K. Prasad and J. G. Chen, J. New Mater. Electrochem. Syst., 2008, 11, 243–251 CAS.
- Y. Wang, S. Song, V. Maragou, P. K. Shen and P. Tsiakaras, Appl. Catal., B, 2009, 89, 223–228 CrossRef CAS.
- Y. Hara, N. Minami, H. Matsumoto and H. Itagaki, Appl. Catal., A, 2007, 332, 289–296 CrossRef CAS.
- Q. Zhu, S. Zhou, X. Wang and S. Dai, J. Power Sources, 2009, 193, 495–500 CrossRef CAS.
- H. Chhina, S. Campbell and O. Kesler, J. Power Sources, 2007, 164, 431–440 CrossRef CAS.
- Z. Shengsheng, Z. Hong, Y. Hongmei, H. Junbo, Y. Baolian and M. Pingwen, Chin. J. Catal., 2007, 28, 109–111 CrossRef.
- Z. J. Mellinger, E. C. Weigert, A. L. Stottlemyer and J. G. Chen, Electrochem. Solid-State Lett., 2008, 11, B63–B67 CrossRef CAS.
- E. C. Weigert, D. V. Esposito and J. G. Chen, J. Power Sources, 2009, 193, 501–506 CrossRef CAS.
- P. K. Shen, S. Yin, Z. Li and C. Chen, Electrochim. Acta, 2010, 55, 7969–7974 CrossRef CAS.
- D. V. Esposito and J. G. Chen, Energy Environ. Sci., 2011, 4, 3900–3912 CAS.
- R. Ganesan, D. J. Ham and J. Lee, Electrochem. Commun., 2007, 9, 2576–2579 CrossRef CAS.
- R. Xu, F. Xu, M. Pan and S. Mu, RSC Adv., 2013, 3, 764–773 RSC.
- N. R. Elezovic, B. M. Babic, P. Ercius, V. R. Radmilovic, L. M. Vracar and N. V. Krstajic, Appl. Catal., B, 2012, 124, 390–397 CrossRef.
- N. R. Elezovic, B. M. Babic, L. Gajić-Krstajic, P. Ercius, V. R. Radmilovic, N. V. Krstajic and L. M. Vračar, Electrochim. Acta, 2012, 69, 239–246 CrossRef CAS.
- M. D. Obradović, S. L. Gojković, N. R. Elezović, P. Ercius, V. R. Radmilović, L. M. Vračar and N. V. Krstajić, J. Electroanal. Chem., 2012, 671, 24–32 CrossRef.
- A. C. Garcia and E. A. Ticianelli, Electrochim. Acta, 2013, 106, 453–459 CrossRef CAS.
- Z. Yan, W. Wei, J. Xie, S. Meng, X. Lü and J. Zhu, J. Power Sources, 2013, 222, 218–224 CrossRef CAS.
- F. Li, H. Gong, Y. Wang, H. Zhang, Y. Wang, S. Liu, S. Wang and C. Sun, J. Mater. Chem. A, 2014, 2, 20154–20163 CAS.
- L. Xiong, L. Zheng, C. Liu, L. Jin, Q. Liu and J. Xu, J. Electrochem. Soc., 2015, 162, F468–F473 CrossRef CAS.
- G. R. Dieckmann and S. H. Langer, Electrochim. Acta, 1998, 44, 437–444 CrossRef CAS.
- L. M. Vracar, S. L. Gojkovic, N. R. Elezovic, V. R. Radmilovic, M. M. Jaksic and N. V. Krstajic, J. New Mater. Electrochem. Syst., 2006, 9, 99–106 CAS.
- E. Slavcheva, V. Nikolova, T. Petkova, E. Lefterova, I. Dragieva, T. Vitanov and E. Budevski, Electrochim. Acta, 2005, 50, 5444–5448 CrossRef CAS.
- K. Sasaki, L. Zhang and R. R. Adzic, Phys. Chem. Chem. Phys., 2008, 10, 159–167 RSC.
- K. W. Park and K. S. Seul, Electrochem. Commun., 2007, 9, 2256–2260 CrossRef CAS.
- N. R. Elezovic, B. M. Babic, L. Gajic-Krstajic, V. Radmilovic, N. V. Krstajic and L. Vracar, J. Power Sources, 2010, 195, 3961–3968 CrossRef CAS.
- N. R. Elezovic, B. M. Babic, V. R. Radmilovic, L. M. Vracar and N. V. Krstajic, Electrochim. Acta, 2011, 56, 9020–9026 CrossRef CAS.
- B. Y. Xia, S. Ding, H. B. Wu, X. Wang and X. Wen (David), RSC Adv., 2012, 2, 792–796 RSC.
- F. Shi, L. R. Baker, A. Hervier, G. A. Somorjai and K. Komvopoulos, Nano Lett., 2013, 13, 4469–4474 CrossRef CAS PubMed.
- A. Kumar and V. Ramani, J. Electrochem. Soc., 2013, 160, F1207–F1215 CrossRef CAS.
- S. Siracusano, A. Stassi, E. Modica, V. Baglio and A. S. Arico, Int. J. Hydrogen Energy, 2013, 38, 11600–11608 CrossRef CAS.
- C. Zhang, H. Yu, Y. Li, L. Fu, Y. Gao, W. Song, Z. Shao and B. Yi, J. Electroanal. Chem., 2013, 701, 14–19 CrossRef CAS.
- K. Tiido, N. Alexeyeva, M. Couillard, C. Bock, B. R. MacDougall and K. Tammeveski, Electrochim. Acta, 2013, 107, 509–517 CrossRef CAS.
- D. Huang, B. Zhang, J. Bai, Y. Zhang, G. Wittstock, M. Wang and Y. Shen, Electrochim. Acta, 2014, 130, 97–103 CrossRef CAS.
- S. Meenakshi, K. G. Nishanth, P. Sridhar and S. Pitchumani, Electrochim. Acta, 2014, 135, 52–59 CrossRef CAS.
- I. Savych, J. Bernard d'Arbigny, S. Subianto, S. Cavaliere, D. J. Jones and J. Rozière, J. Power Sources, 2014, 257, 147–155 CrossRef CAS.
- W. A. Rigdon and X. Huang, J. Power Sources, 2014, 272, 845–859 CrossRef CAS.
- B. Ruiz-Camacho, O. Martínez-Álvarez, H. H. Rodríguez-Santoyo and V. Granados-Alejo, J. Electroanal. Chem., 2014, 725, 19–24 CrossRef CAS.
- K. Jukk, N. Kongi, A. Tarre, A. Rosental, A. B. Treshchalov, J. Kozlova, P. Ritslaid, L. Matisen, V. Sammelselg and K. Tammeveski, J. Electroanal. Chem., 2014, 735, 68–76 CrossRef CAS.
- C. W. Liu, C. M. Lai, J. N. Lin, L. D. Tsai and K. W. Wang, RSC Adv., 2014, 4, 15820–15824 RSC.
- Y. Li, C. Liu, Y. Liu, B. Feng, L. Li, H. Pan, W. Kellogg, D. Higgins and G. Wu, J. Power Sources, 2015, 286, 354–361 CrossRef CAS.
- C. Odetola, L. Trevani and E. Bradley Easton, J. Power Sources, 2015, 294, 254–263 CrossRef CAS.
- X. L. Sui, Z. B. Wang, Y. F. Xia, M. Yang, L. Zhao and D. M. Gu, RSC Adv., 2015, 5, 35518–35523 RSC.
- H. Shintani, Y. Kojima, K. Kakinuma, M. Watanabe and M. Uchida, J. Power Sources, 2015, 294, 292–298 CrossRef CAS.
- N. R. Elezović, B. M. Babić, V. R. Radmilovic, L. M. Vračar and N. V. Krstajić, Appl. Catal., B, 2013, 140–141, 206–212 CrossRef.
- N. R. Elezovic, P. Ercius, J. Kovac, V. R. Radmilovic, B. M. Babic and N. V. Krstajic, J. Electroanal. Chem., 2015, 739, 164–171 CrossRef CAS.
- A. L. Santos, D. Profeti and P. Olivi, Electrochim. Acta, 2005, 50, 2615–2621 CrossRef CAS.
- M. S. Saha, R. Li, M. Cai and X. Sun, Electrochem. Solid-State Lett., 2007, 10, B130–B133 CrossRef CAS.
- K. S. Lee, I. S. Park, Y. H. Cho, D. S. Jung, N. Jung, H. Y. Park and Y. E. Sung, J. Catal., 2008, 258, 143–152 CrossRef CAS.
- F. Takasaki, S. Matsuie, Y. Takabatake, Z. Noda, A. Hayashi, Y. Shiratori, K. Ito and K. Sasaki, J. Electrochem. Soc., 2011, 158, B1270–B1275 CrossRef CAS.
- F. Takasaki, Z. Noda, A. Masao, Y. Shiratori, K. Ito and K. Sasaki, ECS Trans., 2009, 25, 831–837 CAS.
- K. Sasaki, F. Takasaki, Z. Noda, S. Hayashi, Y. Shiratori and K. Ito, ECS Trans., 2010, 33, 473–482 CAS.
- K. Kakinuma, M. Uchida, T. Kamino, H. Uchida and M. Watanabe, Electrochim. Acta, 2011, 56, 2881–2887 CrossRef CAS.
- Y. Fan, J. Liu, H. Lu, P. Huang and D. Xu, Electrochim. Acta, 2012, 76, 475–479 CrossRef CAS.
- M. Dou, M. Hou, D. Liang, W. Lu, Z. Shao and B. Yi, Electrochim. Acta, 2013, 92, 468–473 CrossRef CAS.
- N. R. Elezović, B. M. Babić, V. R. Radmilovic and N. V. Krstajić, J. Electrochem. Soc., 2013, 160, F1151–F1158 CrossRef.
- N. R. Elezovic, V. R. Radmilovic, J. Kovac, B. M. Babic, L. M. Gajic-Krstajic and N. V. Krstajic, RSC Adv., 2015, 5, 15923–15929 RSC.
- M. A. Hoque, D. C. Higgins, F. M. Hassan, J. Y. Choi, M. D. Pritzker and Z. Chen, Electrochim. Acta, 2014, 121, 421–427 CrossRef CAS.
- N. Zhana, S. Zhang, C. Du, Z. Wang, Y. Shao, F. Kong, Y. Lin and G. Yin, Electrochim. Acta, 2014, 117, 413–419 CrossRef.
- T. Tsukatsune, Y. Takabatake, Z. Noda, T. Daio, A. Zaitsu, S. M. Lyth, A. Hayashi and K. Sasaki, J. Electrochem. Soc., 2014, 161, F1208–F1213 CrossRef.
- M. Dou, M. Hou, F. Wang, D. Liang, Q. Zhao, Z. Shao and B. Yi, J. Electrochem. Soc., 2014, 161, F1231–F1236 CrossRef.
- E. Antolini and E. R. Gonzalez, Solid State Ionics, 2009, 180, 746–763 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |