DOI:
10.1039/C5RA22224A
(Paper)
RSC Adv., 2016,
6, 2602-2610
Self-assembled thermosensitive nanoparticles based on oligoethylene glycol dendron conjugated doxorubicin: preparation, and efficient delivery of free doxorubicin
Received
23rd October 2015
, Accepted 20th December 2015
First published on 22nd December 2015
Abstract
An amphiphilic dendron–drug conjugate was synthesized via oligoethylene glycol (OEG) dendrons coupled with anticancer drug doxorubicin (DOX). The amphiphilic conjugate OEG–DOX (GD) self-assembled into spherical nanoparticles in aqueous solution, with mean diameters of approximately 212.5 nm. As an effective drug carrier, DOX was entrapped into GD to form drug-loaded nanoparticles (GD/D) with high drug loading content (24%, wt%), due to the enhanced hydrophobic/aromatic interactions between the DOX moiety in GD and free DOX. Besides, GD nanoparticles exhibited thermo-induced collapse and aggregation, therefore, the temperature-dependent drug release profiles of DOX from GD/D nanoparticles were measured and the possible mechanism was proposed. Moreover, hemolytic activity of GD/D revealed the good blood compatibility, and the cytotoxicity of DOX in GD/D was enhanced significantly in vitro against HepG2 cells. Overall, amphiphilic conjugate GD was considered to be potentially feasible to overcome formulation challenges for drug delivery and to be used in clinic.
1. Introduction
Benefiting from their unique merits, amphiphilic polymers decorated with polyethylene glycol (PEG) have been researched broadly to construct drug delivery systems for hydrophobic anticancer drugs.1–5 These drug delivery systems improve the solubility of hydrophobic drugs, stabilize and protect drugs from degradation, facilitate targeted drug delivery, enhance accumulation in the tumor tissue via the enhanced permeability and retention (EPR) effect, prolong circulation time by avoiding the rapid renal clearance and reticuloendothelial systems (RES) reorganization, and decrease disruption in cellular uptake.6–11 However, the PEGylated strategies present limitations, such as structural heterogeneity due to the polydispersity of PEG chains, low drug loading content (DLC), poor physical stability, drug leakage, and adverse side effects.12–14 To overcome these limitations, oligoethylene glycol (OEG) with well-defined structure and molar mass are synthesized and applied as the drug carriers.15–18
Obviously, it should be more beneficial if the drug could be delivered by a carrier that could respond to physiopathological signals from an underlying disease.19–21 The nanoparticles with thermosensitive feature could undergo a phase transition from an expanded hydrophilic structure below the lower critical solution temperature (LCST) to a compact hydrophobic structure above LCST, which may result in destabilization of the nanoparticles, accumulate in desired position and/or trigger a burst release of the encapsulated drugs.22–24 As the thermosensitive compounds, OEG dendrons with varying LCST are designed and synthesized by Zhang and co-workers.25,26 Based on the excellent water solubility, biocompatibility, nontoxicity, and thermosensitive property, OEG dendrons are utilized to prepare amphiphilic codendrimers which are applied as drug carrier in our previous research.27,28
For physical entrapment, the DLC and release profiles are affected by the interactions between the drug and the hydrophobic segments of amphiphilic polymers, therefore, the composition of the polymers must be considered in the design and evaluation of copolymer-based drug formulations.29–31 According to the excellent compatibility, it is expected that the same or similar structure between the hydrophobic segments of amphiphilic polymers and the entrapped drug could enhance the interactions, result in promoted DLC and controlled release. Park and co-workers demonstrated that doxorubicin (DOX) conjugated with PEG could assist to form and stabilize DOX nanoaggregates due to the interaction between DOX moiety in the conjugate and free DOX, this nanoaggregates showed high DLC (∼25%), enhanced cellular uptake, and increased cytotoxicity against KB cells.32 Besides, DOX was conjugated with PEGylated peptide to encapsulate free DOX successfully by Byun and co-workers.33 Other researches performed by Allen and co-workers indicated utilizing docetaxel (DTX) as the hydrophobic moiety to form PEG–DTX conjugates could entrap free DTX with higher DLC, and the morphologies of these nanoaggregates are influenced by the presence of conjugated DTX in the micelle core.30,34 These researches demonstrated the enhanced DLC and controlled release profiles would be obtained using these amphiphilic carriers with anticancer drug as hydrophobic segment, on account of the strong hydrophobic interactions and excellent compatibility.
Considering the stimuli responsibility and composition, in the work presented here, the amphiphilic OEG–DOX conjugate (GD) is designed as the drug carrier and further loaded DOX by physical entrapment (GD/D). As the drug carrier, the advantages of GD are expected as follows: (1) based on the thermosensitive property, this GD/D delivery system would be stable during the drug transportation in the blood at normal temperature, and could release a large amount of DOX to inhibit the proliferation of cancer cells after arrived at tumor tissue with higher temperature. (2) Utilizing OEG dendron as the hydrophilic segment results amphiphilic molecule GD with well-defined structure and molar mass, which avoids structural heterogeneity in the amphiphilic molecules. (3) GD shows a good capacity to encapsulate the free DOX due to the strong hydrophobic/aromatic interactions between DOX moiety in GD and the free DOX. (4) The anticancer efficacy of GD/D could be enhanced significantly in vitro. For these purposes, the physicochemical characteristics of conjugate GD and GD/D such as solubility, morphology, thermosensibility, cytotoxicity and release profiles are studied to evaluate the great potential of GD as a thermosensitive drug carrier in long-acting interventinal chemotherapy.
2. Experiment part
2.1 Materials
Compound 1 was synthesized according to previous reports.25,26 Tetrahydrofuran (THF) was dried by refluxing with lithium aluminum hydride (LAH) under N2 and distilled just before use. Dichloromethane (DCM) was refluxed with calcium hydride and distilled just before use. Doxorubicin hydrochloride (DOX·HCl, purity > 98%), rhein (RHE, purity > 98%) were purchased from Dalian Meilun Biotech Co., Ltd. (Liaoning, China). Hydroxycamptothecine (HCPT, purity > 98%) was purchased from Aladdin Co. Ltd. (China). Vitamin E (VE, purity > 98%) was purchased from Jiakangyuan Technology Development Co., Ltd. (Beijing, China). Podophyllotoxin (PPT, purity > 98%) was purchased from Nanjing Zelang Medical Technology Co., Ltd. (Jiangsu, China). Other reagents and solvents were purchased as reagent grade and used without further purification. Silica gel 300–400 mesh and Sephadex LH-20 gel were used as the stationary phase for column chromatography.
2.2 Syntheses
Compound 2. LiOH·H2O (0.10 g, 1.64 mmol) was added into a solution of compound 1 (2.00 g, 0.82 mmol) in methanol (20 mL) and water (10 mL) at −5 °C with stirring, and the reaction temperature was allowed to raise to room temperature. After stirring for 3 h, the solvents were evaporated in vacuo at room temperature. The residue was dissolved with DCM, and the solution pH was adjusted carefully with 10% KHSO4 aqueous solution to approximately 5–6. After the organic phase had been washed with brine and dried over MgSO4, the crude product was purified by column chromatography with DCM/MeOH (20/1, v/v) to yield compound 2 as a slight yellow oil (1.90 g, 95%). 1H NMR (CDCl3): δ = 1.17–1.23 (t, 27H, CH3), 3.48–3.50 (m, 18H, CH2), 3.55–3.67 (m, 72H, CH2), 3.70–3.74 (m, 18H, CH2), 3.82–3.86 (m, 30H, CH2), 4.12–4.16 (m, 18H, CH2), 4.18–4.26 (m, 6H, CH2), 4.46–4.48 (s, 6H, CH2), 6.56–6.58 (s, 6H, CH), 7.45 (s, 2H, CH). 13C NMR (CDCl3): δ = 15.10, 66.55, 68.82, 69.05, 69.30, 69.57, 69.71, 69.77, 70.35, 70.47, 70.54, 70.63, 70.65, 70.77, 72.25, 72.48, 73.19, 106.23, 109.38, 125.26, 133.69, 137.86, 144.38, 152.59, 167.07 ppm.
Compound 3. The acid compound 2 (1.90 g, 0.78 mmol) and pentafluorophenol (0.22 g, 1.20 mmol) were dissolved in DCM (30 mL) and stirred for 10 min. DCC (0.37 g, 1.80 mmol) was then added. The mixture was stirred overnight at room temperature before being washed with saturated NaHCO3 and brine successively. After drying over MgSO4, the organic phase was purified by column chromatography with DCM/MeOH (40/1, v/v) to yield a slightly yellow oil (1.50 g, 74%). 1H NMR (CDCl3): δ = 1.19–1.24 (t, 27H, CH3), 3.38–3.46 (m, 18H, CH2), 3.50–3.63 (m, 72H, CH2), 3.68–3.74 (m, 18H, CH2), 3.80–3.86 (m, 30H, CH2), 4.10–4.15 (m, 18H, CH2), 4.23–4.29 (m, 6H, CH2), 4.58–4.62 (s, 6H, CH2), 6.40–6.42 (s, 6H, CH), 7.40 (s, 2H, CH). 13C NMR (CDCl3): δ = 15.10, 61.70, 66.55, 68.82, 69.05, 69.30, 69.57, 69.71, 69.77, 70.35, 70.47, 70.54, 70.63, 70.65, 70.77, 72.25, 72.48, 73.19, 107.23, 110.38, 121.26, 133.69, 137.86, 144.38, 152.59, 162.07 ppm.
OEG-DOX conjugate (GD). The solution of compound 3 (0.40 g, 0.20 mmol) in DMF (10 mL) was added into a solution of DOX·HCl (0.12 g, 0.03 mmol), TEA (67 mg, 0.09 mmol), and DMAP (20 mg) in DMF (10 mL) at −5 °C with stirring, and the reaction temperature was allowed to raise to room temperature. After stirring for 24 h, the solvents were evaporated in vacuo at room temperature, the residue was dissolved with DCM (30 mL). After the organic phase had been washed with 0.2 M HCl solution (30 mL) and dried over MgSO4, the crude product was purified by LH-20 gel column with methanol to afford GD as a red oil (0.31 g, 70%). 1H NMR (CDCl3): δ = 1.19 (t, 27H, CH3), 1.30 (d, 3H, CH3), 1.78–2.01 (m, 2H, CH2), 2.00 (br, 7H, OH, CH2, NH), 2.20 (m, 1H, CH2), 2.35 (d, 1H, CH2), 3.00–3.09 (m, 2H, CH2), 3.25–3.29 (m, 2H, CH), 3.41–3.90 (m, 132H, CH2, CH3), 4.01–4.26 (m, 26H, CH2, CH), 4.30–4.48 (m, 6H, CH2), 4.59–4.68 (m, 3H, CH2, CH), 5.30 (br, 1H, CH), 5.52 (br, 1H, NH), 6.54 (s, 6H, CH), 7.15 (s, 2H, CH), 7.40 (d, 1H, CH), 7.75 (t, 1H, CH), 8.02 (d, 1H, CH); 13C NMR (CDCl3): δ = 14.70, 17.64, 26.81, 35.30, 43.77, 45.50, 56.32, 63.01, 66.15, 80.75, 70.09, 71.03, 72.15, 72.33, 72.51, 72.79, 72.87, 73.29, 73.43, 73.51, 73.62, 73.69, 73.72, 73.52, 73.84, 73.98, 95.00, 105.53, 108.09, 122.19, 123.46, 125.06, 129.39, 130.65, 131.29, 135.48, 145.08, 162.07, 150.81, 187.69 ppm. HRMS: m/z: calcd for C145H231NO60 [M + Na]+ 2970.1697; found 2970.4153.
2.3 Hydrodynamic size measurements
The hydrodynamic sizes of drug-loaded nanoparticles were determined by dynamic light scattering (DLS) using a Zetasizer Nano-ZS analyzer (Malvern Instruments) with an integrated 4 mV He–Ne laser, λ = 633 nm, which used backscattering detection (scattering angle θ = 173°) at different temperature. Samples were prepared by dissolving in deionized water at a concentration of 2 mg mL−1.
2.4 Transmission electron microscopy
Transmission electron microscopy (TEM) measurements were performed with a JEM-1400 instrument at an accelerating voltage of 100 KV. Samples were prepared by drop-casting GD and GD/D solutions (0.2 mg mL−1) onto carbon-coated copper grids, air-drying at room temperature and then dyeing with uranyl acetate.
2.5 Critical aggregation concentration
The CAC of the amphiphilic conjugate GD estimated by a fluorescence spectroscopic method using an F-4500 FL Spectrophotometer and pyrene (Py) as the fluorescence probe. Typically, Py solutions in acetone were added to each eppendorf (EP) tube. The acetone was then evaporated, leaving 6.00 × 10−5 mol of Py in each tube. Aqueous solutions with various concentrations (from 1.0 × 10−3 to 1.0 mg mL−1) were prepared and added to the tubes. The mixtures were sonicated for 2 min and stirred at room temperature for 12 h. The spectroscopy measurements were conducted at an excitation wavelength of 334 nm.
2.6 Thermosensitive property
Turbidity measurements were conducted on a Cary 100 UV-Vis spectrophotometer (Agilent Technologies) equipped with a thermostatically regulated bath. Aqueous compound solutions were placed in the spectrophotometer (path length 1 cm) and heated at a rate of 0.2 °C min−1. The absorption of the solution at λ = 500 nm was recorded. The LCST was determined to be the temperature at which the transmittance at λ = 500 nm reached 50% of its initial value.
2.7 Preparation of drug-loaded nanoparticles
Drug-loaded nanoparticles were prepared by the dialysis method. Briefly, the amphiphilic compound GD (8 mg) and doxorubicin (DOX), hydroxycamptothecine (HCPT), rhein (RHE), podophyllotoxin (PPT), vitamin E (VE) (4 mg) (DOX·HCl neutralized with two molar equivalent of TEA) was dissolved in methanol (2 mL) in a glass vial at room temperature separately. After that, deionized water (5 mL) was added dropwise into these vials under vigorous stirring. The mixture solution was transferred into a dialysis membrane (MWCO 14000) and dialyzed against deionized water (1 L) at room temperature, which was renewed every 4 h for 24 h to remove the organic solvent and the free drug. To quantify the encapsulated drug, 1 mL solutions of the drug-loaded nanoparticles were freeze-dried and then 1 mL MeOH were added, the samples were analyzed by high-performance liquid chromatography (HPLC) using a Dionex Ultimate 3000 with a UV detector at the corresponding wavelength. The quantitative analysis was carried out on a Thermo C18 (4.6 mm × 250 mm, 5 μm), and the sample injection volume was 20 μL. The details for all hydrophobic drugs are shown in Table 1. The entrapment efficiency (EE) and drug loading content (DL) were calculated as follows:
| EE = (weight of loaded drug/weight in feed) × 100% |
| DL = (weight of loaded drug/weight of drug-loaded nanoparticle) × 100% |
Table 1 Conditions for hydrophobic drugs measured by HPLC
| Sample |
λ (nm) |
Eluent |
Flow rate (mL) |
Regression equation |
R2 |
| DOX |
478 |
KH2PO4 (2.5%)/CH3OH 3/7 |
0.8 |
y = 0.26x + 0.01 |
0.9999 |
| PPT |
292 |
CH3CN/H3PO4 (0.1%) 4/6 |
1.0 |
y = 0.18x − 0.02 |
0.9999 |
| VE |
285 |
CH3OH |
1.0 |
y = 0.05x − 0.30 |
0.9999 |
2.8 Drug release in vitro
The DOX-loaded nanoparticles (GD/D) aqueous solution (2 mL) was transferred into a dialysis membrane (MWCO 14000), immersed in 50 mL of PBS solutions. The release studies were performed at 37 and 40 °C respectively with continuous magnetic stirring at 100 rpm. At predetermined time intervals, 2 mL of the external solution was withdrawn and replenished with an equal volume of fresh media for analysis. The drug release study was performed for 84 h. The amount of released DOX was analyzed with HPLC using a Dionex Ultimate 3000 with a UV detector operated at 478 nm. The release experiments were conducted in triplicate, and the results were presented as the mean values plus the standard deviation (±SD).
2.9 Hemolytic activity
Approximately 5 mL of blood was taken from the ear artery of a male New Zealand white rabbit and centrifuged at 3000 rpm for 5 min. The plasma supernatant was removed, and the erythrocytes were re-suspended in normal saline solution. The red blood cells (RBC) were again centrifuged at 3000 rpm for 5 min to collect packed RBC. The GD/D solutions were incubated with the 2% (v/v) RBC suspension at 37 °C for 3 h with different concentration. The unlysed RBCs were removed by centrifugation, 100 μL of the supernatant was pipetted into a 96-microwell plate, and the absorbance was measured at 540 nm using a microplate reader (Versamax Tunable Microplate Reader). The results were expressed as the percentages of hemolysis with the assumption that deionized water caused 100% hemolysis and 150 mM NaCl solution caused 0% hemolysis. Each experiment was done in triple. The data were shown as the mean values plus standard deviation (±SD). All experimental procedures were performed in accordance with the Guidelines for Ethical and Regulatory for Animal Experiments as defined by the Institutional Animal Care and Use Committee of Peking University Health Science Center.
2.10 MTT assay
The cytotoxicity against the human liver cancer cell line (HepG2 cells) was assessed by methyl thiazolyl tetrazolium (MTT) assay. Briefly, cells were cultured in DMEM medium supplemented with 10% fetal calf serum, 1% L-glutamine, and 0.1 mg mL−1 streptomycin at 37 °C with 5% CO2 and then seeded in 96-well plates at a density of 5000 cells per well. The GD, GD/D, and free DOX solution were added into the wells. After incubation for 48 h at 37 °C with 5% CO2, the growth medium was replaced with fresh DMEM. Then, 10 μL MTT solution was added to each well and incubation was continued for another 4 h. The medium was removed and 200 μL DMSO was added into each well to dissolve the formazan, assisted by pipetting in and out several times. The absorbance of the solution in each well was measured using an ELISA plate reader at a test wavelength of 570 nm to determine the OD value. The cell inhibition rate was calculated as follows. Cell inhibition = (1 − ODtreated/ODcontrol) × 100%, where ODtreated was obtained from the cells treated by the nanoparticles or free DOX, ODcontrol was obtained for the cells treated by the culture medium, and the other culture conditions were the same. Each experiment was performed in quintuplicate. The data were shown as the mean values plus the standard deviation (±SD).
2.11 Statistical analysis
The results obtained from hydrodynamic diameter, drug-loading content, release experiments, and cytotoxicity were expressed as the mean ± standard deviation (SD). Statistical analysis was performed with SPSS 19.0 software. Student's t-test was used to evaluate the differences between groups, and P < 0.05 was considered statistically significant.
2.12 Instrumentation and measurements
1H NMR and 13C NMR spectra were recorded on a Bruker AV 600 spectrometers with CDCl3 as solvent, and chemical shifts were reported as δ values (ppm) relative to internal Me4Si. Molecular weight was determined by Autoflex III MALDI-TOF mass spectrophotometer with ESI detector. FT-IR spectra were recorded on a FTIR-8400S (Shimadzu Japan) spectrometer. Samples were pressed into potassium bromide (KBr) pellets.
3. Results and discussion
3.1 Synthesis and characteristics of OEG–DOX conjugate
To construct the amphiphilic conjugate, the oligoethylene glycol (OEG) dendron active ester was prepared, and the synthetic route of OEG dendron conjugated DOX (GD) is shown in Scheme 1. The compounds 1, 2 and 3 were synthesized following the procedures in previous reports. DOX was conjugated with active ester 3 through the amide bond, after purification by Sephadex LH-20 column chromatography with methanol as eluent, amphiphilic conjugate GD was obtained with a yield of 70%. The resultant compound GD displayed a good solubility in common solvent, such as MeOH, THF, DCM, DMF, and water, due to the excellent solubility of the OEG dendron.
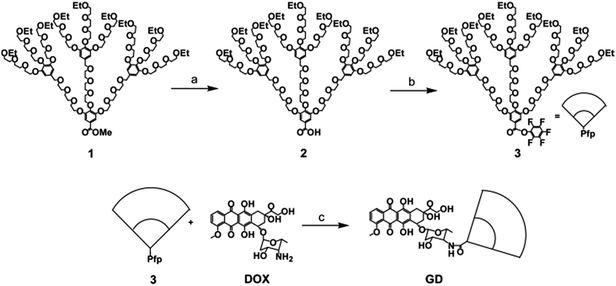 |
| | Scheme 1 Synthetic route of the amphiphilic conjugate GD. Reagents and conditions: (a) LiOH, MeOH, −5 to 25 °C, 3 h (95%); (b) Pfp, DCC, DCM, 25 °C, 12 h (74%); (c) DOX, TEA, DMAP, DMF, −5 to 25 °C, 24 h (70%). Pfp = pentafluorophenol, DCC = dicyclohexylcarbodiimide, DCM = dichloromethane, TEA = triethylamine, DMAP = N,N-dimethylaminopyridine, DMF = dimethylformamide. | |
The structure of GD was confirmed by 1H NMR spectroscopy, and the signals are ascribed separately and shown in Fig. 1. Comparing with the compound 3, the 1H NMR spectrum of GD presented the characteristic peaks of DOX; the signals at 8.0–7.3 and 5.3 ppm were attributed to the benzene ring protons and tetrahydropyran ring protons separately. The signals around 1.1 and 1.2 ppm corresponded to the proton resonance of methyl terminals of OEG dendron and DOX respectively, the integral ratio of these two signals was 9.0. These results suggested the successful synthesis of the amphiphilic compound GD. The proposed structure of GD is further verified by the FT-IR technique, meanwhile the DOX and compound 1 are measured for comparing (Fig. 2). FT-IR results also confirmed the generation of amphiphilic compound GD based on the characteristic stretching vibration of carbonyl absorption at 1648 cm−1 from amide group, the typical stretching C–O–C absorption at 1110 cm−1 from OEG dendron, and the strong stretching N–H absorption at 3440 cm−1 from DOX. Then, the DOX content within GD (17.3%, wt%) was determined by UV-HPLC at 478 nm, which was consistent with the theoretical value (17.1%). All these results proved that amphiphilic conjugate GD had been successfully synthesized via amide coupling reaction. Benefiting from its amphiphilic structure with hydrophilic OEG and hydrophobic DOX, the self-assembly of GD into nanoparticles can be expected in aqueous solution to encapsulate the hydrophobic anticancer drugs into the core by means of hydrophobic interaction, which is beneficial for further biomedical applications.
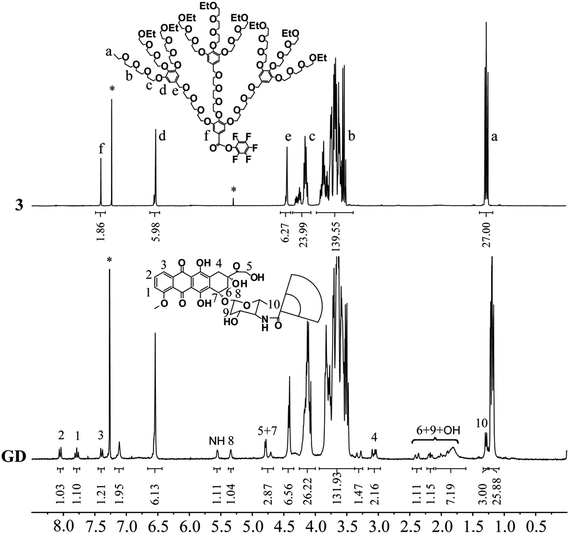 |
| | Fig. 1 1H NMR spectra of compound 3 and GD in CDCl3. | |
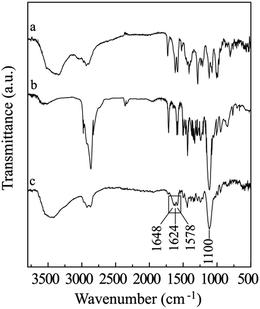 |
| | Fig. 2 FT-IR spectra of DOX (a), compound 1 (b), and GD (c). Samples were pressed into potassium bromide pellets. | |
3.2 Self-assemble behavior
Due to the excellent solubility, amphiphilic conjugate GD was dissolved in deionized water directly with concentration of 2 mg mL−1 and self-assembled into the nanoparticles in the aqueous solution, the particle size was detected by DLS (Fig. 3a). It was found that GD formed nanoparticles with the mean hydrodynamic diameter of approximately 212.5 ± 11.1 nm (Table 2). TEM micrograph revealed that GD could aggregate into spherical nanoparticles with the mean diameter of approximately 180.2 ± 3.1 nm (Fig. 3b). The diameter of GD nanoparticles observed by TEM is smaller than that obtained from the DLS measurement. The size of the nanoparticles measured by DLS reflects the hydrodynamic diameter and the TEM image shows that of dried particles, which is most likely due to shrinkage of the OEG portion.31,35
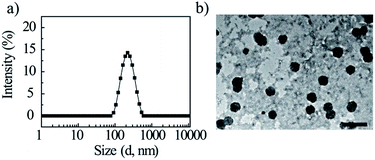 |
| | Fig. 3 DLS curves of amphiphilic compound GD in aqueous solution (a); TEM image of self-assembled aggregates (b). Scale bar: 500 nm. | |
Table 2 Results of the drug-loaded nanoparticlesa
| Samples |
Dhb (nm) |
ζc (mV) |
Diameterd (nm) |
DLCe (%) |
| Prepared by dialysis method. Dh: hydrodynamic diameter, 2 mg mL−1, DLS detected, n = 3. ζ: zeta potential, 2 mg mL−1, DLS detected, n = 3. The average diameters of 20 particles in TEM images. Detected by UV-HPLC. Not detected. |
| GD |
212.5 ± 11.1 |
−28.1 |
180.2 ± 3.1 |
0 |
| GD/D |
240.5 ± 16.7 |
−26.7 |
210.4 ± 2.7 |
24.7 ± 4.1 |
| GD/P |
252.1 ± 4.4 |
−21.2 |
n.d.f |
12.3 ± 1.2 |
| GD/V |
238.8 ± 0.8 |
−27.2 |
n.d. |
6.5 ± 0.4 |
The aggregation behavior of amphiphilic compound GD was further characterized by fluorescence spectroscopy using pyrene as a probe, which was sensitive to the surrounding environment, and the ratio of the intensity of the third (384 nm) and first (374 nm) peaks was used to confirm the CAC. It was apparent that the ratio of I384/I374 remained constant below the CAC and then changed substantially, and the CAC was detected as 17.4 μg mL−1 (Fig. 4).
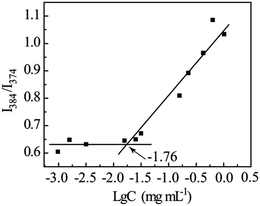 |
| | Fig. 4 Dependence of intensity ratio I384/I374 as a function of amphiphilic compound GD concentration. | |
3.3 Thermosensitive behavior
Owing to the presence of OEG dendron, the GD nanoparticles were assessed to have thermosensitive properties in aqueous solution and undergo a phase transition from homogeneous solution to heterogeneous aggregates around a lower critical solution temperature (LCST). UV-vis spectrophotometry and dynamic light scattering (DLS) technology were thus applied to investigate the thermosensitive behavior of GD and to determine its apparent LCST. The turbidity curve measured from 33 to 45 °C is plotted in Fig. 5a. Compound GD showed typical thermosensitive behavior, and its LCST is 39.2 °C. The thermally induced aggregation was further investigated by DLS to record the temperature-dependent aggregate size, the diameter curve measured from 33 to 42 °C is plotted in Fig. 5b. The hydrodynamic diameters (Dh) of GD nanoparticles (2 mg mL−1) were measured to increase from 200 nm at 33 °C to 2800 nm at 42 °C, and the appearance of GD solution was changed from fully transparent to turbid. The increase in Dh could be attributed to the internanoparticle aggregation. It is supposed that the novel amphiphilic conjugate is of great potential use as smart drug carriers, which is stable under physiological condition and aggregates at the tumor tissue induced by slight higher temperature to increase the cellular uptake due to the hydrophobic interaction.36,37
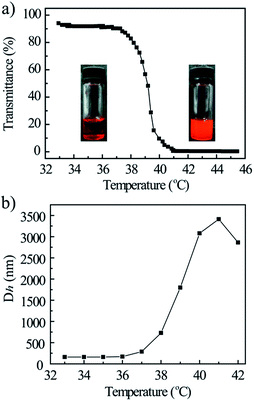 |
| | Fig. 5 Plots of the transmittance (a) and Dh (b) vs. temperature for 0.2 wt% GD aqueous solution. | |
3.4 Drug-loading capacity
To assess the influence of amphiphilic compound structure on drug incorporation, doxorubicin (DOX), hydroxycamptothecine (HCPT), rhein (RHE), podophyllotoxin (PPT), and vitamin E (VE) were utilized as the model drugs to prepare drug-loaded nanoparticles (Fig. 6). DOX·HCl was first neutralized by excess triethylamine to remove the hydrochloride, and then these drugs were incubated with GD in DMF, dialyzed against deionized water to remove DMF and the free drugs.
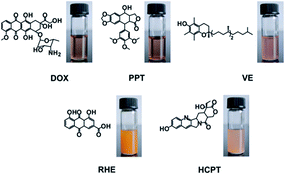 |
| | Fig. 6 Structures of hydrophobic drugs and images of drug-loaded nanoparticle solutions. | |
Among the selected model drugs, DOX could be encapsulated in amphiphilic GD successfully, and the drug-loading content (DLC) was 24.7% (physical entrapment, Table 2), the DOX-loaded nanoparticles (GD/D) solution was fully transparent red solution (Fig. 6). RHE and HCPT formed precipitation after dialysis, although PPT and VE could be entrapped into GD, the DLC of PPT-loaded nanoparticles (GD/P) and VE-loaded nanoparticles (GD/V) were 12.3% and 6.5% separately (Table 2), significant lower than that of GD/D. It seems that GD presented the excellent DOX-loading capacity, which could be explained by the stronger hydrophobic interactions induced by the similar structure between the hydrophobic segments of GD molecules and the free DOX.38,39 Besides, it was reported that DOX having an anthraycline ring structure forms a dimmer in an aqueous solution due to π–π interaction between the planar, aromatic anthracycline rings.32 Thus, DOX could be entrapped into the GD nanoparticles by the hydrophobic and aromatic interactions between the DOX moiety in GD and the free DOX; meanwhile, excellent drug-loading capacity could be achieved.
3.5 Characterization of DOX-loaded nanoparticles
The particle size of GD/D nanoparticles with a concentration of 2 mg mL−1 in aqueous solution were investigated by DLS, and the mean diameter of approximately 240.5 ± 16.7 nm (Table 2). The stability of nanoparticles solution was investigated by monitoring for nanoparticles size changes using DLS (Fig. 7a). It was apparant that the particle sizes of DOX-loaded nanoparticles were approximately 240 nm for 10 days incubation at 37 °C. These results suggested that GD/D nanoparticles had the favorable stability in aqueous solution, which was likely due to the OEG dendron surface.
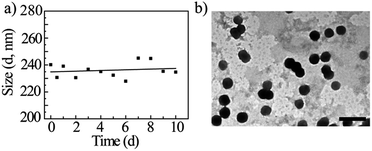 |
| | Fig. 7 Mean GD/D diameter as a function of time in deionized water (a), and TEM micrograph (b). Scale bar: 500 nm. | |
The TEM micrograph revealed that GD/D nanoparticles were all spherical with the mean diameter of approximately 210.4 ± 2.7 nm (Fig. 7b). Similar as GD nanoparticles, the diameter of GD/D nanoparticles observed by TEM is smaller than that obtained from the DLS measurement. In addition, it was found that the sizes of DOX-loaded nanoparticles were larger than the blank nanoparticles, indicating that the DOX was successfully loaded into the core of GD nanoparticles due to the hydrophobic/aromatic interactions between the conjugated and the free DOX.40
3.6 Release behavior of thermosensitive DOX-loaded nanoparticles
After drug-loaded GD/D nanoparticles were prepared, the DOX release profiles from the thermosensitive nanoparticles were evaluated under a simulated physiological condition (PBS, pH 7.4) at 37 °C and in an acidic environment (PBS, pH 5.5 and 6.8) at 37 °C and 40 °C in separate trials to assess the feasibility of using GD/D nanoparticles as an anticancer drug delivery system, as tumor tissues have a more acidic environment and slight higher temperature than normal physiological conditions.41–44 A control experiment using free DOX was also performed in pH 5.5 PBS at 37 °C, all the release results are shown in Fig. 8. For free DOX, the burst release of up to 70% within the initial 2 h was observed, and then followed by the complete release within 10 h. The release of DOX from GD/D nanoparticles at physiological temperature (37 °C) showed the pH-dependent property. Approximately 80% of DOX was released from the nanoparticles at 84 h post-dialysis in PBS 5.5, while only 39% and 32% of DOX was released in PBS 6.8 and 7.4 respectively, since the solubility of DOX was enhanced significantly in acidic aqueous solution. Although DOX could be released at the extracellular environment of tumor tissues, a large amount of DOX (>60%) was retained inside the nanoparticles. Furthermore, under the tumor microenvironment (pH 5.5), the DOX release rate was much faster, with approximately profiles were shown to be temperature dependent. At 37 °C (<LCST), approximately 80% of the loaded DOX was sustained release from the GD/D nanoparticles for 84 h, which was most likely due to the slow diffusion of DOX in the nanoparticles. At 40 °C (>LCST), the release of DOX was enhanced dramatically on account of the temperature-induced structural changes of the nanoparticles, approximately 50% of DOX was released within the initial 4 h followed by another slow release procedure (about 40%, from 4 to 84 h). The OEG dendron shell of GD/D nanoparticles becomes hydrophobic above the LCST, which could lead to the partial deformation of nanoparticles resulting in the faster drug release.45–47 These results prompted that the GD/D delivery system would be stable during the drug transportation in the blood, after arrived at tumor tissue with higher temperature and acidic environment, it could release a large amount of DOX to inhibit the proliferation of cancer cells.
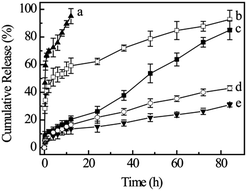 |
| | Fig. 8 Cumulative DOX release in different conditions: free DOX in pH 5.5 PBS at 37 °C (a), GD/D in pH 5.5 PBS at 40 °C (b), GD/D in pH 5.5 PBS at 37 °C (c), GD/D in pH 6.8 PBS at 37 °C (d), and GD/D in pH 7.4 PBS at 37 °C (e) (n = 3). | |
3.7 Cytotoxicity
The hemolytic effect of GD/D on rabbit RBC was investigated to verify the suitability via intravenous administration. The hemolysis rates of GD/D were recorded as a function of concentration, ranging from 0.1 to 20 mg mL−1. After incubation with the 2% (w/v) RBC suspension at 37 °C for 3 h, the hemolysis rates of GD/D solutions was below 5%, suggesting no RBC membrane damage and the good blood compatibility.48,49
The cytotoxicities of GD, GD/D against a liver cancer line (HepG2 cells) were studied using an MTT assay, with the concentration ranging from 0.1 to 100 μg mL−1 (DOX equivalent concentration) (Fig. 9). The IC50 values for free DOX, GD, and GD/D were 27.1, 45.6, and 8.0 μg mL−1 (DOX equivalent concentration) respectively after incubation for 48 h. Compared with free DOX, after conjugation of DOX with OEG dendron showed significant decrease in cytotoxicity due to the shielding effect (p < 0.001), which could reduce severe side effects of DOX. GD/D exerted a higher cytotoxicity effect against HepG2 cells at the same dose (p < 0.001), which suggested much more DOX was transferred into the tumor cells and proved the activity of GD/D nanoparticles was greater than that of the free drug. The enhanced cytotoxicity of GD/D nanoparticles could be attributed to the facilitated endocytotic transport and quick intracellular DOX release within the endosomes, relative to passive diffusion of free doxorubicin through the cell membrane.15,50 This was probably due to the mechanism of endocytosis that might effectively circumvent the multi-drug resistance (MDR) effect that occurs for free DOX.51,52 In the previous study of DOX-loaded nanoparticles, the similar enhanced uptake of DOX against HepG2 cells was observed.53 It was expected that GD/D nanoparticles could augment the therapeutic effect and reduce the severe side effects of DOX.33
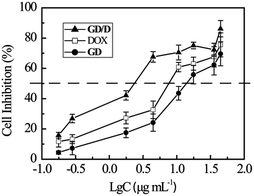 |
| | Fig. 9 Cytotoxicities of DOX, GD, and GD/D nanoparticles against HepG2 cells for 48 h incubation (n = 5). | |
4. Conclusions
Anticancer drug DOX was conjugated with the hydrophilic OEG dendron to form the amphiphilic thermosensitive conjugate GD (LCST 39.2 °C), which could self-assemble into spherical nanoparticles in aqueous solution with the mean diameter of approximately 212 nm. The amphiphilic GD was utilized to entrap DOX (GD/D) and the drug-loading content was approximately 24%, due to the strong hydrophobic/aromatic interactions between DOX moiety in GD and the free DOX. The release of DOX from GD/D nanoparticles was enhanced dramatically on account of the temperature-induced structural changes of the amphiphilic dendron GD, which could maximize the anticancer efficacy of drug delivery system. The drug-loaded GD/D nanoparticles presented good blood compatibility (hemolysis rate < 5%), and the cytotoxicity was enhanced significantly in vitro via MTT assay against HepG2 cells. Based on these advanced properties, including high drug-loading content, temperature-dependent drug release, and higher anticancer efficacy, it seems that amphiphilic conjugate with anticancer drug as hydrophobic segment and thermosensitive dendron as hydrophilic segment hold great promise for efficient delivery and release of relative anticancer drugs.
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 21444003) and PUMC Youth Fund (No. 33320140184).
Notes and references
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- X.-B. Xiong, Z. Binkhathlan, O. Molavi and A. Lavasanifar, Acta Biomater., 2012, 8, 2017–2033 CrossRef CAS PubMed.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- D. Mishra, J. R. Hubenak and A. B. Mathur, J. Biomed. Mater. Res., Part A, 2013, 101, 3646–3660 CrossRef PubMed.
- K. Park, ACS Nano, 2013, 7, 7442–7447 CrossRef CAS PubMed.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- M. J. Joralemon, S. McRae and T. Emrick, Chem. Commun., 2010, 46, 1377–1393 RSC.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- J. Khandare, M. Calderon, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- A. Vergara, L. Paduano, V. Vitagliano and R. Sartorio, Phys. Chem. Chem. Phys., 1999, 1, 5377–5383 RSC.
- F. M. Veronese, O. Schiavon, G. Pasut, R. Mendichi, L. Andersson, A. Tsirk, J. Ford, G. Wu, S. Kneller, J. Davies and R. Duncan, Bioconjugate Chem., 2005, 16, 775–784 CrossRef CAS PubMed.
- V. P. Torchilin, Pharm. Res., 2007, 24, 1–16 CrossRef CAS PubMed.
- W. Wang, J. Ding, C. Xiao, Z. Tang, D. Li, J. Chen, X. Zhuang and X. Chen, Biomacromolecules, 2011, 12, 2466–2474 CrossRef CAS PubMed.
- N. Sadhukhan, T. Muraoka, M. Ui, S. Nagatoishi, K. Tsumoto and K. Kinbara, Chem. Commun., 2015, 51, 8457–8460 RSC.
- L. Yu, Z. Zheng, Y. Liu, Z. Li and X. Wang, RSC Adv., 2015, 5, 64832–64840 RSC.
- Y. Shen, E. Jin, B. Zhang, C. J. Murphy, M. Sui, J. Zhao, J. Wang, J. Tang, M. Fan, E. van Kirk and W. J. Murdoch, J. Am. Chem. Soc., 2010, 132, 4259–4265 CrossRef CAS PubMed.
- J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- B. Daglar, E. Ozgur, M. E. Corman, L. Uzun and G. B. Demirel, RSC Adv., 2014, 4, 48639–48659 RSC.
- O. J. Cayre, N. Chagneux and S. Biggs, Soft Matter, 2011, 7, 2211–2234 RSC.
- X. Wu, X. He, L. Zhong, S. Lin, D. Wang, X. Zhu and D. Yan, J. Mater. Chem., 2011, 21, 13611–13620 RSC.
- J. Akimoto, M. Nakayama, K. Sakai and T. Okano, Biomacromolecules, 2009, 10, 1331–1336 CrossRef CAS PubMed.
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840–853 CrossRef CAS PubMed.
- W. Li, A. Zhang, Y. Chen, K. Feldman, H. Wu and A. D. Schluter, Chem. Commun., 2008, 5948–5950 RSC.
- W. Li, A. Zhang and A. D. Schluter, Chem. Commun., 2008, 5523–5525 RSC.
- Y. Guo, Y. Zhao, J. Zhao, M. Han, A. Zhang and X. Wang, Bioconjugate Chem., 2014, 25, 24–31 CrossRef CAS PubMed.
- Y. Guo, Y. Zhao, M. Han, C. Hao and X. Wang, J. Mater. Chem. B, 2013, 1, 6078–6084 RSC.
- K. Letchford, R. Liggins and H. Burt, J. Pharm. Sci., 2008, 97, 1179–1190 CrossRef CAS PubMed.
- A. S. Mikhail and C. Allen, Biomacromolecules, 2010, 11, 1273–1280 CrossRef CAS.
- H. Sun, B. Guo, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2009, 30, 6358–6366 CrossRef CAS PubMed.
- H. S. Yoo and T. G. Park, J. Controlled Release, 2004, 100, 247–256 CrossRef CAS.
- G. Y. Lee, K. Park, S. Y. Kim and Y. Byun, Eur. J. Pharm. Biopharm., 2007, 67, 646–654 CrossRef CAS PubMed.
- J. Liu, P. Zahedi, F. Zeng and C. Allen, J. Pharm. Sci., 2008, 97, 3274–3290 CrossRef CAS PubMed.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 5757–5766 CrossRef CAS PubMed.
- H. Akita, T. Nakatani, K. Kuroki, K. Maenaka, K. Tange, Y. Nakai and H. Harashima, Int. J. Pharm., 2015, 490, 142–145 CrossRef CAS PubMed.
- Y. Li, G. H. Gao and D. S. Lee, Adv. Healthcare Mater., 2013, 2, 388–417 CrossRef CAS PubMed.
- W. Zhang, Y. Li, L. Liu, Q. Sun, X. Shuai, W. Zhu and Y. Chen, Biomacromolecules, 2010, 11, 1331–1338 CrossRef CAS PubMed.
- S. Lv, W. Song, Z. Tang, M. Li, H. Yu, H. Hong and X. Chen, Mol. Pharmaceutics, 2014, 11, 1562–1574 CrossRef CAS PubMed.
- C. Y. Zhang, Y. Q. Yang, T. X. Huang, B. Zhao, X. D. Guo, J. F. Wang and L. J. Zhang, Biomaterials, 2012, 33, 6273–6283 CrossRef CAS.
- J. Cao, T. Su, L. Zhang, R. Liu, G. Wang, B. He and Z. Gu, Int. J. Pharm., 2014, 471, 28–36 CrossRef CAS PubMed.
- K. P. Koutroumanis, R. G. Holdich and S. Georgiadou, Int. J. Pharm., 2013, 455, 5–13 CrossRef CAS PubMed.
- T. Yahara, T. Koga, S. Yoshida, S. Nakagawa, H. Deguchi and K. Shirouzu, Surg. Today, 2003, 33, 243–248 CrossRef PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- X. Wan, T. Liu and S. Liu, Biomacromolecules, 2011, 12, 1146–1154 CrossRef CAS PubMed.
- S. J. T. Rezaei, M. R. Nabid, H. Niknejad and A. A. Entezami, Int. J. Pharm., 2012, 437, 70–79 CrossRef CAS PubMed.
- X. Wang, S. Li, Z. Wan, Z. Quan and Q. Tan, Int. J. Pharm., 2014, 463, 81–88 CrossRef CAS PubMed.
- F. le Dévédec, S. Strandman, P. Hildgen, G. Leclair and X. X. Zhu, Mol. Pharm., 2013, 10, 3057–3066 CrossRef.
- Z. Zhao, M. He, L. Yin, J. Bao, L. Shi, B. Wang, C. Tang and C. Yin, Biomacromolecules, 2009, 10, 565–572 CrossRef CAS PubMed.
- F.-Q. Hu, X.-L. Wu, Y.-Z. Du, J. You and H. Yuan, Eur. J. Pharm. Biopharm., 2008, 69, 117–125 CrossRef CAS PubMed.
- H. S. Yoo, E. A. Lee and T. G. Park, J. Controlled Release, 2002, 82, 17–27 CrossRef CAS PubMed.
- R. Tang, W. Ji, D. Panus, R. N. Palumbo and C. Wang, J. Controlled Release, 2011, 151, 18–27 CrossRef CAS PubMed.
- H. S. Yoo, K. H. Lee, J. E. Oh and T. G. Park, J. Controlled Release, 2000, 68, 419–431 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 









