
Iron-containing platinum-based catalysts as cathode and anode materials for low-temperature acidic fuel cells: a review
Abstract
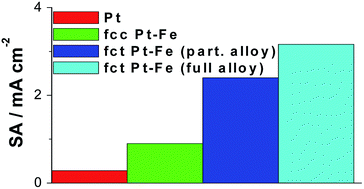
The high availability and low cost of Fe make it an interesting element for use in non-precious Pt-free catalysts and Pt-based catalysts for low-temperature fuel cells. Pt–Fe compounds can present three crystal structures, these are a disordered fcc PtxFe alloy and two ordered intermetallic alloys (fcc Pt3Fe and fct PtFe types). Fe-containing Pt-based binary and ternary catalysts in the different crystal structures have been tested both as anode and cathode materials in low-temperature acid fuel cells. In this work an overview of the application of Fe-containing catalysts as cathode materials for oxygen reduction and as anode materials for methanol and ethanol oxidation in low-temperature polymer electrolyte fuel cells fuelled with hydrogen or low molecular weight alcohols, is presented. Moreover, the stability of iron in Pt-based binary and ternary catalysts towards dissolution in acid medium is discussed.
 Please wait while we load your content...
Please wait while we load your content...

