
Diastereoselective synthesis of 3,3-dimethylazetidines via an intramolecular iodine-mediated cyclization reaction†
Hai-Shan Jin‡
,
Rui Sun‡,
Wei Zhou and
Li-Ming Zhao*
School of Chemistry and Chemical Engineering, and Jiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, Jiangsu Normal University, Xuzhou 221116, Jiangsu, China. E-mail: lmzhao@jsnu.edu.cn
First published on 28th January 2016
Abstract
A number of bioactive molecules possess a 3,3-dimethylazetidine moiety. An iodine-mediated intramolecular cyclization reaction of γ-prenylated amines has been developed to provide a convenient route to 3,3-dimethylazetidines in a highly diastereoselective manner. The presented approach not only offers a new strategy for the synthesis of the bioactively important 3,3-dimethylazetidines but also provides an opportunity to extend the application of γ-prenylated amine synthons in organic synthesis.
Azetidines are found in many pharmaceutical compounds and natural products.1 More specifically, 3,3-dimethylazetidines are endowed with interesting biological activities, such as being dopamine receptor antagonists, acetylcholine receptor agonists, and PDE4 inhibitors (Fig. 1).2 A particularly interesting structural feature of this scaffold is the gem-dimethyl group at C3. The presence of the gem-dimethyl moiety in bioactive compounds was realized to improve their biological properties, such as the hydrophobic interaction with the receptor sites of bioactive molecules.3 This is particularly relevant in pharmaceutical and biomedical research since the introduction of such a fragment into biomolecules could influence either the pharmacodynamic or the pharmacokinetic properties of a drug candidate. About 10% of all approved drugs contain at least one gem-dimethyl group, thus highlighting its relevance in the discovery of new drugs.4 Moreover, the gem-dimethyl fragment is a common substructure present in natural products,5 which serve as important sources for drug discovery. However, in contrast to the well-established methods for the synthesis of azetidines,6 the synthesis of 3,3-dimethylazetidines is still an undeveloped area, as shown by the limited literature on the subject.7
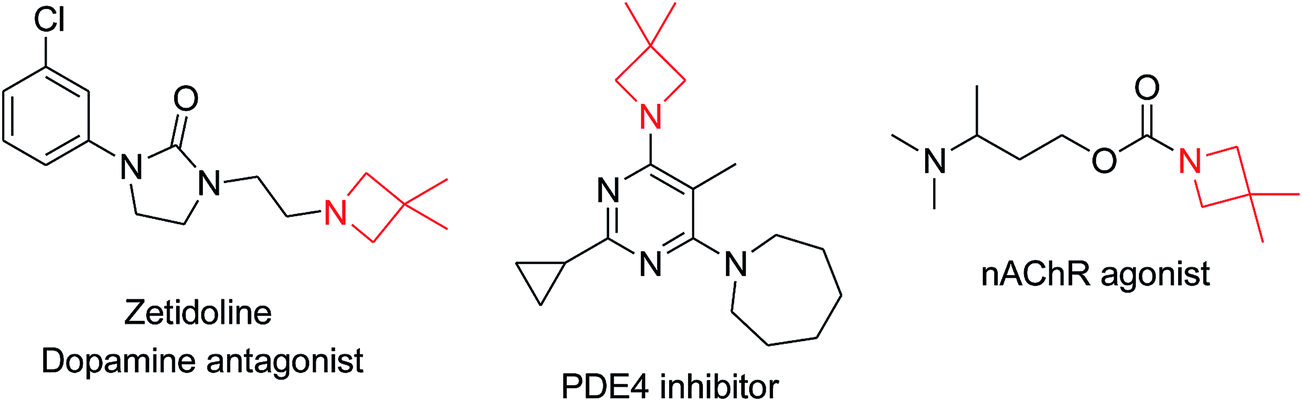 | ||
| Fig. 1 Selected examples of biologically relevant 3,3-dimethylazetidine derivatives. | ||
Our interest in 3,3-dimethylazedines grew out of our studies on prenylation in recent years. In the past few years, our group has been involved in the development of methodologies towards simple and efficient regioselective prenylation for the synthesis of various α-prenylated compounds.8 Due to their unusual structures, which contain two methyl groups in C1 or C3 position (α-prenylated compounds or γ-prenylated compounds), prenylated compounds are of considerable importance for the regioselective allylation and have been used as building blocks for the preparation of otherwise inaccessible compounds. In this regard, we have reported the synthesis of substituted tetrahydrofurans through Prins cyclization and the synthesis of furo[3,2-c]benzopyrans via intramolecular [4 + 2] cycloaddition reaction using α-prenylated compounds, respectively.9 Recently, we were interested in the chemistry of the isomeric γ-prenylated compounds, which can be prepared easily according to a reported procedure.8a We considered whether we might be able to use γ-prenylated derivatives in order to synthesize the otherwise inaccessible compounds, such as 3,3-dimethylazedines. To the best of our knowledge, the direct transformation of γ-prenylated amines to 3,3-dimethylazedines has not yet been disclosed. Herein, we wish to report our study on this subject.
γ-Prenylated amine 1a was employed as a model compound for the optimization of the reaction conditions (Table 1). Synthesis of various nitrogen heterocycles utilizing iodoamination has received considerable attention because such reactions leave an iodine atom attached to the target molecule that is amenable to further transformation.10 We started our investigations using the conditions which had been applied in the iodocyclization of homoallylic amines.11 As expected, the cyclized product 2a was obtained in 59% yield when commercially available acetonitrile was used as solvent in the presence NaHCO3 and iodine (entry 1). Although the procedure was effective, the conversion of the starting material was not satisfactory enough and long reaction times were required. Gratifyingly, a significant improvement was noticed when the solvent CH3CN was dried resulting in a shortened reaction time for the preparation of the corresponding product (entry 2). It seemed that the use of an anhydrous solvent was essential to achieve the cyclization efficiently. Scaling down the base load from 5.0 equiv. to 3.0 equiv. gave no substantial decrease in the yield (entry 3). Next, the effects of other bases were investigated. Somewhat surprisingly the yield of the reaction decreases on increasing the strength of the base. For example, changing the base from NaHCO3 to the slightly stronger K2CO3 resulted in the formation of the product 2a in 35% yield (entry 4). In the case of KOH, only small amounts (10%) of the desired cyclized product 2a were formed (entry 5). Since NaHCO3 has two recognition possibilities with the transition state: by the hydrogen atom (electrophilic assistance) or by the oxygen (nucleophilic assistance), to verify the actual role of NaHCO3 in the reaction, we examined the cyclization reactions of 1a in the presence of other bases such as NaOAc and Na3PO4.12 Both reactions were completed within 6 h to give the product 2a in good yields (entries 6 and 7). These results clearly show the basic role of NaHCO3 for promoting the present cyclization reaction. Without NaHCO3, a low yield was observed, suggesting its critical role in the completion of the reaction (entry 8). Thus, the optimal reaction conditions we established involved the use of 1a (1.0 equiv.), NaHCO3 (3.0 equiv.), and I2 (3.0 equiv.) in 15 mL of CH3CN at room temperature for 6 h. The structure of compound 2a was unambiguously confirmed by X-ray crystallography (see the ESI†).
|
|||||
|---|---|---|---|---|---|
| Entry | Base (equiv.) | Solvent | t (h) | 2aa (%) | Recovery of 1a (%) |
| a Isolated yield. | |||||
| 1 | NaHCO3 (5.0) | CH3CN | 10 | 59 | 20 |
| 2 | NaHCO3 (5.0) | Dry CH3CN | 6 | 92 | — |
| 3 | NaHCO3 (3.0) | Dry CH3CN | 6 | 90 | — |
| 4 | K2CO3 (3.0) | Dry CH3CN | 6 | 35 | — |
| 5 | KOH (3.0) | Dry CH3CN | 6 | 10 | 70 |
| 6 | NaOAc (3.0) | Dry CH3CN | 6 | 84 | — |
| 7 | Na3PO4 (3.0) | Dry CH3CN | 6 | 82 | — |
| 8 | — | Dry CH3CN | 12 | 44 | 52 |
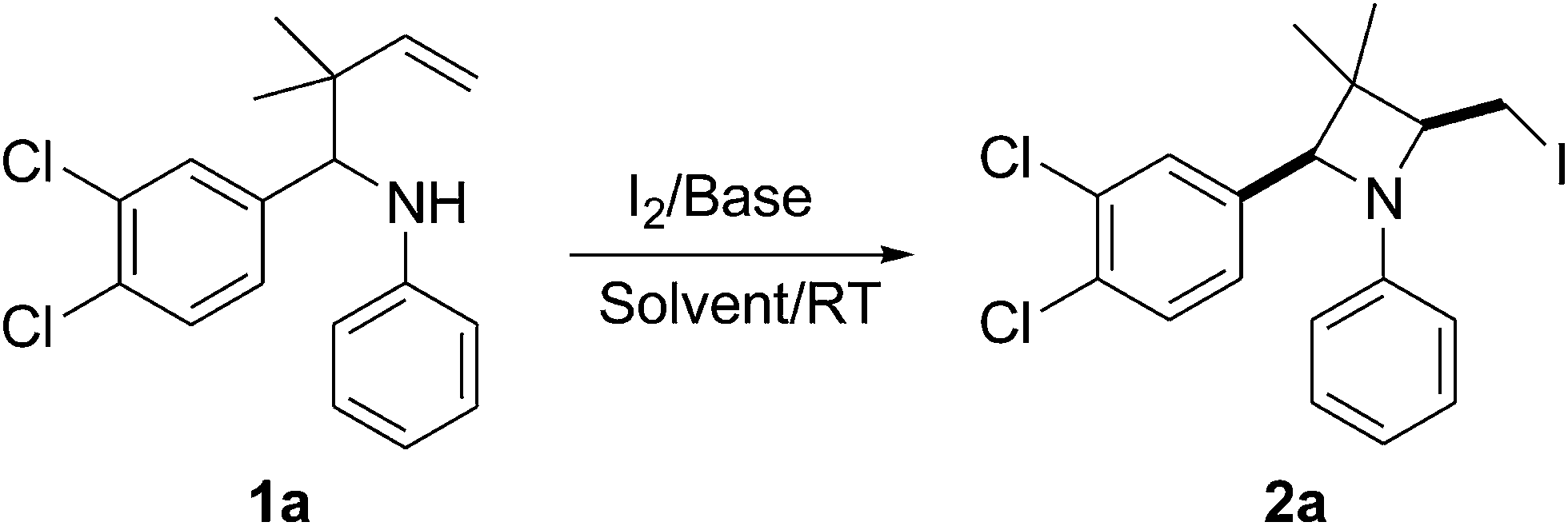
Having identified suitable reaction conditions, we further investigated the scope of the reaction with a variety of substrates. As summarized in Table 2, the protocol could be used for the synthesis of diverse 3,3-dimethylazedines and all of the reactions could proceed smoothly, giving the desired products in good yields. It seemed that the nature of the substituent on the two aromatic rings (i.e., Ar1 and Ar2), whether electron-donating or electron-withdrawing, has no significant influence on the reactions. Furthermore, the position of the substituent on the aromatic rings practically does not affect product yield. However, from Table 2, we can see that the substrates containing two electron-withdrawing groups (chloro) on the aromatic rings worked better, affording the corresponding cyclized products 2k-m in excellent yields (entries 10–12). This is beneficial for further transformations and future applications because the ability of halide to modify and/or enhance the biological properties of compounds is of great interest in medicinal chemistry. It is worth noting that excellent diastereoselectivities were obtained for a selection of electron-donating and -withdrawing substituents. Under the optimized reaction conditions, the reactions proceeded without any complications, affording the expected products 2b-t in diastereomerically pure form in all cases. The high diastereoselectivities indicate that the application of this reaction provides a very efficient diastereoselective synthetic method for the preparation of 3,3-dimethylazedines.
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | Ar1, Ar2 | 2 | Yielda (%) | drb |
| a Isolated yield.b The ratio was determined by 1H NMR. | |||||
| 1 | 1b | Ph, 4-ClC6H4 | 2b | 86 | >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | 1c | Ph, 4-BrC6H4 | 2c | 69 | >99:1 |
| 3 | 1d | 4-ClC6H4, Ph | 2d | 70 | >99:1 |
| 4 | 1e | 3-ClC6H4, 4-BrC6H4 | 2e | 89 | >99:1 |
| 5 | 1f | 4-BrC6H4, 2-FC6H4 | 2f | 80 | >99:1 |
| 6 | 1g | 4-BrC6H4, 4-ClC6H4 | 2g | 73 | >99:1 |
| 7 | 1h | 4-BrC6H4, 4-BrC6H4 | 2h | 72 | >99:1 |
| 8 | 1i | 4-FC6H4, 4-BrC6H4 | 2i | 69 | >99:1 |
| 9 | 1j | 4-CF3C6H4, 4-BrC6H4 | 2j | 92 | >99:1 |
| 10 | 1k | 3,4-Cl2C6H3, 4-BrC6H4 | 2k | 91 | >99:1 |
| 11 | 1l | 4-BrC6H4, 3,4-Cl2C6H3 | 2l | 85 | >99:1 |
| 12 | 1m | 2,6-Cl2C6H3, 3,4-Cl2C6H3 | 2m | 95 | >99:1 |
| 13 | 1n | 4-FC6H4, 4-MeC6H4 | 2n | 65 | >99:1 |
| 14 | 1o | 2-BrC6H4, 4-MeC6H4 | 2o | 65 | >99:1 |
| 15 | 1p | 4-CF3C6H4, 4-MeC6H4 | 2p | 76 | >99:1 |
| 16 | 1q | 4-MeC6H4, 2-FC6H4 | 2q | 66 | >99:1 |
| 17 | 1r | 4-MeC6H4, Ph | 2r | 86 | >99:1 |
| 18 | 1s | 4-MeOC6H4, 4-ClC6H4 | 2s | 77 | >99:1 |
| 19 | 1t | 4-MeC6H4, 4-MeC6H4 | 2t | 84 | >99:1 |
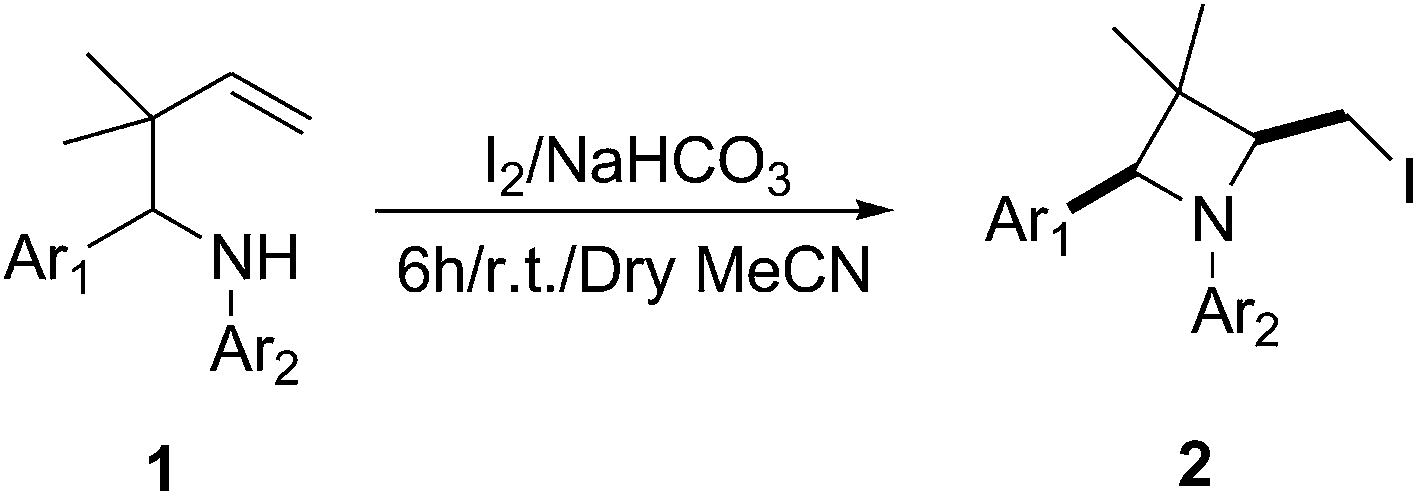
The mechanism of the reaction for the formation of cis-azetidine products 2 can be explained as illustrated in Scheme 1. The process presumably starts with the formation of the two diastereomeric iodiranium species I and II, which originate from the addition of iodine to the C–C double bond. Then, I and II would undergo the elimination of proton under the assistance of NaHCO3 to produce intermediates III and IV, respectively. NaHCO3 can act as either an acid (donate a proton) or a base (accept a proton), however, we have demonstrated the basic role of NaHCO3 in the reaction through several control experiments (Table 1, entries 6 and 7). Then the reaction would proceed through an intramolecular nucleophilic attack of III and IV on the iodiranium. Consequently, two possible pathways resulting in the transition state A (TS-A) and transition state B (TS-B) should be considered. In TS-A, the protons appear in pseudo axial positions and as a result the repulsion between them is not significant. Conversely, in TS-B, the iodiranium is positioned pseudoaxially, leading to increased 1,3-diaxial interaction. On the basis of this analysis, the TS-A from I would be more favorable than the TS-B from II. So the formation of trans-2 is prevented. The TS-A thus formed would lead to the formation of the four-membered azetidine 2 as a cis product.
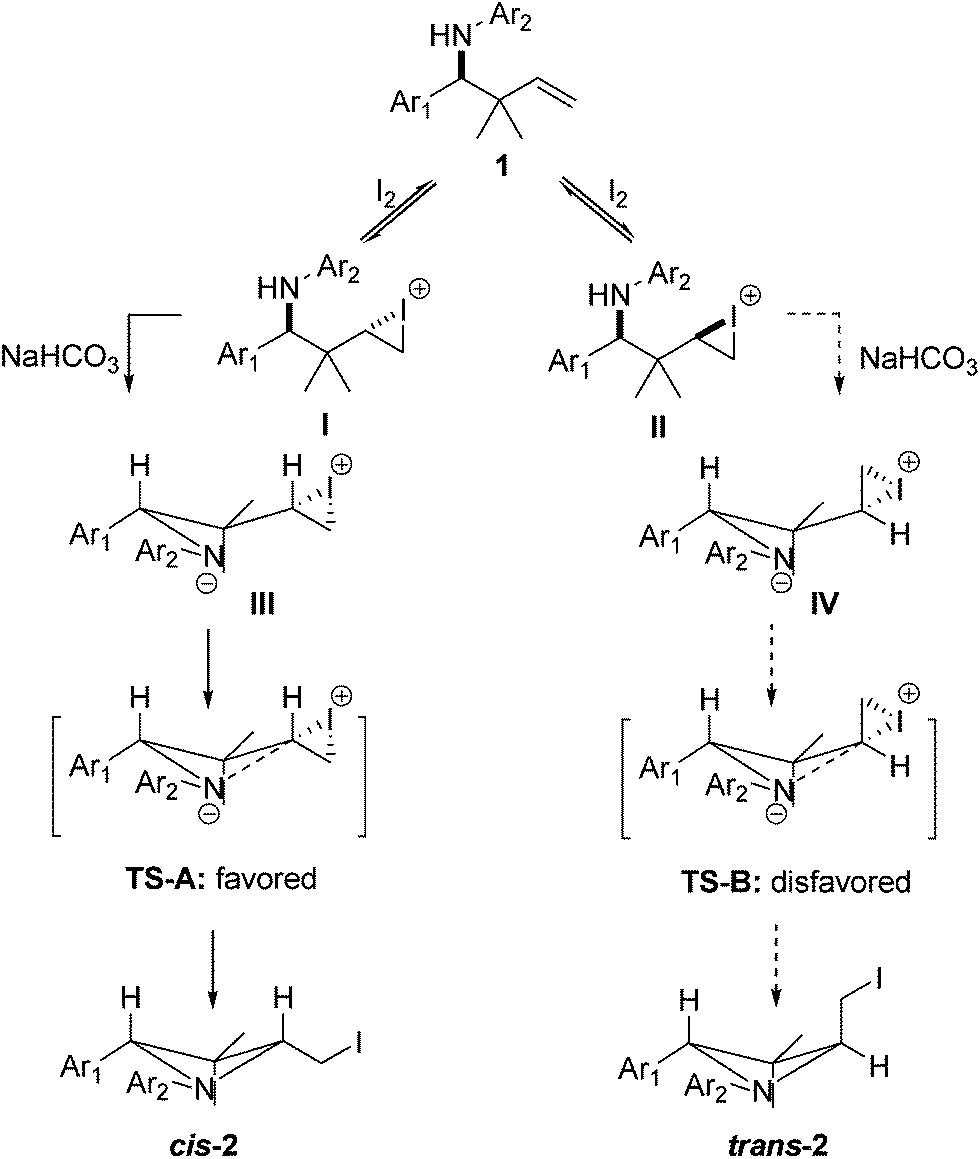 | ||
| Scheme 1 Possible mechanism. | ||
In summary, we have successfully developed a mild and efficient method for the synthesis of 3,3-dimethylazetidines via intramolecular iodine-mediated cyclization reaction from γ-prenylated amines in good yields with high diastereoselectivity. The presented approach not only offers a new strategy for the synthesis of the bioactive important 3,3-dimethylazetidines but also provides an opportunity to extend the application of γ-prenylated amines synthons in organic synthesis. Considering the potential application of this kind of products, we believe that this new methodology may find wide application in both organic and medicinal chemistry.
Acknowledgements
We are thankful for financial support from Major Program of Natural Science Foundation (12KJA150005) and PAPD of Jiangsu Higher Education Institutions, and Innovation Training Program for the Jiangsu College Students (201513988003Y).References
- For reviews, see:
(a) A. R. Pinder, Nat. Prod. Rep., 1992, 9, 491 RSC
. For selected examples, see: (b) J. Frigola, J. Pares, J. Corbera, D. Vano, R. Merce, A. Torrens, J. Mas and E. Valenti, J. Med. Chem., 1993, 36, 801 CrossRef CAS PubMed
-
(a) A. Assandri, G. Galliani, L. Zerilli, G. Tuan, G. Tarzia and D. Barone, Biochem. Pharmacol., 1986, 35, 1459 CrossRef CAS PubMed
-
(a) S. Manabe, H. Takayanagi and C. Nishino, J. Chem. Ecol., 1983, 9, 533 CrossRef CAS PubMed
- J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Mueller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052 CrossRef CAS PubMed
- For recent examples, see:
(a) J. Li, L. Pan, Y. Deng, U. Munoz-Acuna, C. Yuan, H. Lai, H. Chai, T. E. Chagwedera, N. R. Farnsworth and E. J. Carcache de Blanco, J. Org. Chem., 2013, 78, 10166 CrossRef CAS PubMed
- For reviews, see:
(a) C. O. Puentes and V. Kouznetsov, J. Heterocycl. Chem., 2002, 39, 595 CrossRef CAS
-
(a) A. G. J. Anderson and M. T. Wills, J. Org. Chem., 1968, 33, 2123 CrossRef CAS
-
(a) L.-M. Zhao, H.-S. Jin, L.-J. Wan and L.-M. Zhang, J. Org. Chem., 2011, 76, 1831 CrossRef CAS PubMed
-
(a) L.-M. Zhao, F. Dou, R. Sun and A.-L. Zhang, Synlett, 2014, 25, 1431 CrossRef
- For a review, see:
(a) M. J. Mphahlele, Molecules, 2009, 14, 4814 CrossRef CAS PubMed
-
(a) A. Feula, L. Male and J. S. Fossey, Org. Lett., 2010, 12, 5044 CrossRef CAS PubMed
- We thank the reviewer for the valuable input on the manuscript.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1057046. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra21872a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |