
Facile synthesis of well-shaped spinel LiNi0.5Mn1.5O4 nanoparticles as cathode materials for lithium ion batteries†
Yi Cai‡
a,
Shao-Zhuan Huang‡a,
Fa-Shuang Shea,
Jing Liua,
Run-Lin Zhanga,
Zhen-Hong Huanga,
Feng-Yun Wang*b and
Hong-En Wang*a
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan, 430070, China. E-mail: hongenwang@whut.edu.cn; hongen.wang@gmail.com; Fax: +86 2787879468; Tel: +86 2787855322
bThe Cultivation Base for State Key Laboratory, Qingdao University, Qingdao, 266071, China. E-mail: fywang@qdu.edu.cn
First published on 23rd December 2015
Abstract
Spinel LiNi0.5Mn1.5O4 (LNMO) nanoparticles with well-defined polyhedral shapes and mean sizes of ca. 200 nm have been synthesized via a solid-state route using α-MnO2 nanowires as reaction precursors. A structural reorganization is believed to be responsible for the morphology evolution from tetragonal α-MnO2 nanowires to spinel LNMO nanoparticles. Galvanostatic charge–discharge measurements indicate the LNMO nanoparticles exhibit a high initial discharge capacity of 129 mA h g−1 with an 88% capacity retention over 100 cycles at 1C (147 mA h g−1), superior to those of LNMO nanorod counterparts (116 mA h g−1). The superior electrochemical performance of LNMO nanoparticles can be ascribed to their narrow particle size distribution, less particle aggregation, intimate interparticle contact, increased electrical conductivity and lithium ion insertion–extraction kinetics due to the existence of oxygen deficiency and exposed {111} crystal facets.
1. Introduction
Lithium ion batteries (LIBs) have been considered as the next generation energy storage devices for a variety of daily applications such as cellular phones, laptop computers, electric vehicles, etc.1 To further increase the energy density of LIBs, various positive electrode materials have been intensively investigated to obtain either higher working voltage or larger capacity.2–4 Among them, spinel LiMn2O4 is generally considered as an excellent alternative cathode material to commercially layered LiCoO2 because of its low cost, safety, and environmental friendliness.5–8 However, LiMn2O4 has a relatively low operation voltage and suffers from capacity loss due to the Mn3+ dissolution and Jahn–Teller effect.9 In comparison, spinel LiNi0.5Mn1.5O4 (LNMO) is considered as an advanced cathode material for LIBs and has attracted tremendous research attention because of its higher operation voltage (∼4.7 V) and high intrinsic rate capability.10–12 The high operation voltage of LNMO endows the batteries with higher output energy density when using it as cathode to assemble full lithium ion batteries.In principle, LNMO has two different crystal structures:13,14 one adopts a disordered face-centered cubic spinel (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) structure and the other owns an ordered primitive simple cubic crystal symmetry (P4332). For the former, the Li+ ions are located in the Wyckoff 8a sites of the structure, and the Mn4+ and Ni2+ ions are randomly distributed in the 16d sites while the O2− ions occupy the 32e sites. For the latter, the Ni2+, Mn4+, and Li+ ions occupy the 4a, 12d, and 8c sites, respectively, whereas the O2− ions reside in the 8c and 24e sites. It has been established that the preparation of either of the LNMO phases via a solid-state reaction is mainly determined by the annealing conditions.15 For instance, ordered spinel can be synthesized at calcination temperatures around 700 °C and disordered ones are obtained at firing temperatures higher than 700 °C.16,17 The overall reaction upon high firing temperatures can be formulated as LiNi0.5Mn1.5O4 ↔ αLixNi1−xO + βLiNi0.5−yMn1.5+yO4−x + γO2. Based on this equation, it is evident that trace LixNi1−xO is usually formed as an impurity phase during the preparation of non-stoichiometric disordered LNMO materials.18 The electrochemical performance of spinel LNMO is related to several factors: (1) the presence of Mn3+ ions and the degree of cation disorder,19 (2) doping or substitution with cations,20,21 and (3) presence of LiyNi1−yO impurity.22 Therefore, it remains challenging to simultaneously achieve remarkable rate capability and cyclability for LNMO due to complex performance-influencing factors and electrode/electrolyte interface instability under high potentials. Cation doping, surface modifications,23,24 and creating nanostructures can stabilize electrode structure and enhance the formation of passivating solid-electrolyte interphase (SEI) layer. Nonetheless, only limited types of LNMO nanostructures such as nanoparticles,25 hollow spheres,26 porous nanorods,3 core–shell structured porous spheres,27 have been reported so far. In addition, it is not easy to achieve size and shape control of LNMO materials with high purity due to the undesired phase separation and grain sintering in its synthesis involving prolonged calcinations at high temperatures.
m) structure and the other owns an ordered primitive simple cubic crystal symmetry (P4332). For the former, the Li+ ions are located in the Wyckoff 8a sites of the structure, and the Mn4+ and Ni2+ ions are randomly distributed in the 16d sites while the O2− ions occupy the 32e sites. For the latter, the Ni2+, Mn4+, and Li+ ions occupy the 4a, 12d, and 8c sites, respectively, whereas the O2− ions reside in the 8c and 24e sites. It has been established that the preparation of either of the LNMO phases via a solid-state reaction is mainly determined by the annealing conditions.15 For instance, ordered spinel can be synthesized at calcination temperatures around 700 °C and disordered ones are obtained at firing temperatures higher than 700 °C.16,17 The overall reaction upon high firing temperatures can be formulated as LiNi0.5Mn1.5O4 ↔ αLixNi1−xO + βLiNi0.5−yMn1.5+yO4−x + γO2. Based on this equation, it is evident that trace LixNi1−xO is usually formed as an impurity phase during the preparation of non-stoichiometric disordered LNMO materials.18 The electrochemical performance of spinel LNMO is related to several factors: (1) the presence of Mn3+ ions and the degree of cation disorder,19 (2) doping or substitution with cations,20,21 and (3) presence of LiyNi1−yO impurity.22 Therefore, it remains challenging to simultaneously achieve remarkable rate capability and cyclability for LNMO due to complex performance-influencing factors and electrode/electrolyte interface instability under high potentials. Cation doping, surface modifications,23,24 and creating nanostructures can stabilize electrode structure and enhance the formation of passivating solid-electrolyte interphase (SEI) layer. Nonetheless, only limited types of LNMO nanostructures such as nanoparticles,25 hollow spheres,26 porous nanorods,3 core–shell structured porous spheres,27 have been reported so far. In addition, it is not easy to achieve size and shape control of LNMO materials with high purity due to the undesired phase separation and grain sintering in its synthesis involving prolonged calcinations at high temperatures.
In this work, we report the facile synthesis of spinel LNMO nanoparticles via a simple solid-state reaction route using α-MnO2 nanowires as a self-sacrificial template. The LNMO nanoparticles show well-defined polyhedral shapes and have narrow particle size distribution. Different from the mature synthetic routes developed for preparation of various one-dimensional spinel LiMn2O4 nanostructures,9,28–30 the α-MnO2 nanowires favour the formation of polyhedral LNMO nanoparticles during the solid-state lithiation process. The effects of annealing time and starting precursors on the structure and morphology of LNMO have also been investigated systematically. Due to the unique structure and geometry characteristics, the well-shaped LNMO nanoparticles present much higher Li storage capacity as well as better rate performance than those of LNMO nanorods counterparts prepared using β-MnO2 nanorods as precursors.
2. Experimental
2.1 Materials synthesis
The synthesis of α-MnO2 nanowires referred to a published route31 with some revisions. In a typical procedure, 8 mmol Mn(CH3COO)2 and equivalent (NH4)2S2O8 were dissolved in 40 mL de-ionized water, followed by adding 40 mL 0.375 M (NH4)2SO4 solution and stirring for 30 min. The resulting solution was transferred to a 100 mL Teflon-lined stainless steel autoclave and maintained at 120 °C for 12 h. After the reaction, the products were washed with de-ionized water and ethanol for three times. The as-obtained α-MnO2 nanowires were subsequently dried at 80 °C in air for 12 h.For comparison, β-MnO2 nanorods were synthesized by a similar hydrothermal reaction process described above but without adding (NH4)2SO4. In addition, α-MnO2 nanorods were synthesized via hydrothermal reaction of a mixed aqueous solution of 80 mL containing 0.395 g KMnO4 and 0.151 g MnSO4 at 120 °C for 12 h, referring a literature method with some modifications.32
To prepare LNMO nanostructures, a high-temperature solid-state reaction was adopted using LiOH, Ni(NO3)2 and α-MnO2 nanowires (α-MnO2 or β-MnO2 nanorods) with a molar ratio of 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5:1.5 as the starting materials. The mixture was dispersed in absolute ethanol and continuously stirring until the ethanol was evaporated completely. Then the mixture was annealed in a box furnace at 750 °C in air for 10 h with a temperature ramping rate of 2 °C min−1 and finally cooled to room temperature naturally.
:0.5:1.5 as the starting materials. The mixture was dispersed in absolute ethanol and continuously stirring until the ethanol was evaporated completely. Then the mixture was annealed in a box furnace at 750 °C in air for 10 h with a temperature ramping rate of 2 °C min−1 and finally cooled to room temperature naturally.
2.2 Physical characterizations
Crystal structures of the as-prepared samples were identified using a Bruker X-ray diffractometer (XRD) with Cu Kα radiation (λ = 1.54056 Å). The surface morphologies and particle sizes of the as-fabricated materials were observed with a Hitachi S-4800 field-emission scanning electron microscope (FESEM). Transmission electron microscope (TEM) and high resolution transmission electron microscopy (HRTEM) images were acquired on a JEOL JEM-2100F electron microscope with an acceleration voltage of 200 kV. Energy-dispersive X-ray spectroscopy (EDS) was performed using an EDAX Genesis instrument with an acceleration voltage of 30 kV. The surface electronic states of Mn were analyzed by X-ray photoelectron spectroscope (XPS, Thermo fisher, Alpha). Raman spectra were carried out at room temperature, and the signals were recorded by an Invia Raman Microscope (Invia Microscope, Renishaw, UK) using a 633 nm excitation wavelength.2.3 Electrochemical measurements
The working electrodes for lithium-half cells were prepared by thoroughly blending a slurry containing 70 wt% active material (LNMO), 20 wt% conducting carbon black, and 10 wt% polyvinylidene fluoride (PVDF) binder in N-methyl-2-pyrrolidone (NMP). The slurry was uniformly coated on an aluminum foil by a doctor-blade method and then dried at 120 °C in a vacuum oven for 12 h. Coin-type (CR2025) lithium-half cells were assembled in an argon-filled glovebox using lithium foils as counter electrode and reference electrode, 1 M LiPF6 dissolved in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1 v/v) as electrolyte. Cyclic voltammetry (CV) measurements were carried out using a CHI 604E electrochemical workstation at a scan rate of 0.2 mV s−1. Galvanostatic charge–discharge cycling was recorded on a LAND battery testing system (LAND CT2001A, Wuhan) in a potential range of 3.5–5 V (vs. Li+/Li) under different current rates (where 1C was defined as 147 mA g−1). Electrochemical impedance spectra (EIS) were measured with an electrochemical workstation (Autolab PGSTAT 302N) in the frequency range from 100 kHz to 10 mHz. All the electrochemical tests were carried out at room temperature.
3. Results and discussion
3.1 Morphology and structure characterizations of the MnO2 precursors and LiNi0.5Mn1.5O4 products
Spinel LiNi0.5Mn1.5O4 (LNMO) nanostructures were synthesized via a convenient solid-state reaction as schematically illustrated in Fig. 1. In the first step, uniform α-MnO2 nanowires were synthesized by a hydrothermal method and employed as the precursors. Next, Li salt and Ni salt were adsorbed onto the particle surfaces of α-MnO2 nanowires by impregnation and evaporation. Finally, LNMO nanoparticles (NPs) were obtained via high temperature annealing in air. In comparison, LNMO nanorods (NRs) were obtained by a similar solid-state reaction process using β-MnO2 NRs as the precursor. | ||
| Fig. 1 Schematic illustration of the synthetic process of the LNMO nanostructures. | ||
Crystal structures and morphologies of the MnO2 precursors were studied by XRD and SEM characterizations. As shown in the lower XRD pattern of Fig. 2a, all the diffraction peaks of the MnO2 sample obtained by a hydrothermal reaction with the addition of (NH4)2SO4 can be well indexed to the pure tetragonal α-MnO2 phase with I4/m (87) space group (JCPDS card no. 44-0141).30,33 In contrast, the diffraction peaks of the MnO2 sample prepared in the absence of (NH4)2SO4 (the upper XRD pattern of Fig. 2a) can be indexed to tetragonal β-MnO2 with P42/mnm (136) space group (JCPDS card no. 24-0735).34 No peaks for other kinds of manganese oxides phases can be detected, suggesting the relatively high purity of the as-prepared MnO2 products. SEM image shown in Fig. 2b reveals that the as-synthesized α-MnO2 sample comprises a large number of thin nanowires interwoven together. A magnified view (Fig. 2c) discloses that the nanowires have diameters of 20 nm on average and lengths up to several micrometers. These nanowires are bent and cross-linked into a three-dimensional porous interconnected mesh-like structure, which can facilitate the surface adsorption of lithium ions and nickel ions for the subsequent solid-state synthesis of LNMO NPs. In contrast, the as-obtained β-MnO2 product mainly consists of a large number of nanorods with particle diameters of 30–90 nm and lengths up to 1 μm, which form some bundles with side faces fusing together (Fig. 2d).
 | ||
| Fig. 2 (a) XRD patterns of the hydrothermally synthesized MnO2 products; SEM images of (b and c) α-MnO2 nanowires sample, and (d) β-MnO2 nanorods sample. | ||
It is noted that the redox reaction between Mn2+ and S2O82− ions can be formulated as Mn2+ + S2O82− + 2H2O → MnO2 + 2SO42− + 4H+, which includes two half-reactions: (1) Mn2+ + 2H2O → MnO2 + 4H+ + 2e− (standard redox potential E01 = 1.23 V), and (2) S2O82− + 2e− → 2SO42− (E02 = 2.01 V). Based on the difference of the standard redox potential (E0) values of these two half-reactions, the standard Gibbs free energy change (ΔG0) could be estimated to be ca. −151 kJ mol−1, suggesting a strong reaction tendency from a thermodynamics point of view. However, we noticed that this reaction proceeded slowly at room temperature without using catalyst, which was possibly due to its slow reaction kinetics.35,36 In addition, the discrepancy in the crystallographic forms (α-MnO2 and β-MnO2) lies in the existence of additional NH4+ (or K+) ions, which can be trapped in the 2 × 2 tunnels of α-MnO2 and stabilize the larger channel structures.37 The morphology difference between the two MnO2 samples can also be attributed to the possible effects caused by the addition of (NH4)2SO4: (1) alteration of ionic strength during the hydrothermal growth process; (2) selective adsorption of SO42− anions on specific crystallographic facets, which promotes the one-dimensional oriented crystal growth behavior.
After lithiation via solid-state reactions, spinel LNMO nanostructures can be obtained. Fig. 3 shows the XRD patterns of the as-synthesized LNMO nanostructures via solid-state reaction routes using α-MnO2 nanowires and β-MnO2 nanorods as precursors, respectively. In Fig. 3, the dominant diffraction peaks of both two LNMO samples can be indexed to cubic spinel phase of LiNi0.5Mn1.5O4 (JCPDS card no. 80-2162, space group Fdm).10,13,27 No peaks of α-MnO2 or β-MnO2 phases can be detected anymore, revealing the MnO2 precursors have been completely converted into LNMO phase during the solid-state reaction process. In addition, a few minor residual diffraction peaks can be weakly seen and readily indexed to trace rocksalt phase LixNi1−xO,6,15,27 which has been reported to be an impurity occasionally formed during the synthesis of LNMO together with the formation of oxygen vacancies occurring concomitantly. Compared to the standard JCPDS cards, the diffraction peaks of (311), (400), and (440) facets in Fig. 3a are highly intensified, suggesting the possible existence of preferentially exposed crystallographic facets in this sample.
 | ||
| Fig. 3 XRD patterns of the (a) LNMO-NPs and (b) LNMO-NRs. | ||
The morphology of the as-prepared LNMO samples was further observed by SEM and TEM. As displayed in Fig. 4a and S1a,† the LNMO product mainly comprises a large number of nanoparticles with well-defined polyhedral shapes (sharp corners and edges), which is distinct from the one-dimensional structure of the parent α-MnO2 nanowires precursors. The average particle size was estimated to be around 200 nm. Some particles take octahedral appearance and others present truncated octahedral shapes. TEM micrograph shown in Fig. 4b reveals an octahedral LNMO nanoparticle has coarse grain surfaces, which might be beneficial for Li ion insertion/extraction. Fig. 4c and d further depict the TEM and HRTEM micrographs and corresponding fast Fourier transform (FFT) pattern of a truncated octahedral particle, revealing its outer surface is mainly enveloped by {111} crystal facets. This result coincides with recent theoretical studies that lattice planes with low surface energies are more prone to be formed after high temperature calcinations for long time.38 In addition, the well-dispersed LNMO NPs with narrow particle size distribution easily cluster together and form some larger porous secondary-particles, as shown in Fig. S2a.† Energy-dispersive X-ray spectroscopy (EDS) pattern and elemental mapping (Fig. 3 and S2b–d†) further evidence the uniform distribution of O, Mn, and Ni elements throughout the LNMO sample. In comparison, SEM images (Fig. 5a and S1b†) show that the LNMO sample derived from β-MnO2 precursors retains rod-like morphology with larger particle diameter of ∼100 nm and shorter rod length of ∼550 nm on average. TEM micrograph in Fig. 5b depicts the LNMO nanorod has smooth grain surface with sharp tips, differing from that of the LNMO NPs.
 | ||
| Fig. 4 (a) SEM image, (b and c) TEM and (d) HRTEM micrographs of LNMO NPs (inset of panel (d) is a fast Fourier transform pattern taken from HRTEM image). | ||
 | ||
| Fig. 5 (a) SEM image and (b) TEM micrograph of LNMO nanorods. | ||
Raman spectroscopy was further recorded to identify the phase structure, local chemical bonding and cation ordering information in the LNMO samples. As shown in Fig. 6, the strong and wide peaks around 630 cm−1 can be assigned to the symmetric Mn–O stretching vibration (A1g) mode of MnO6 octahedra. The shoulder bands at about 590 cm−1 and 490 cm−1 are assigned to the F(1)2g and F(2)2g species.39 Meanwhile, two weak bands can also be observed at about 403 cm−1 and 390 cm−1 corresponding to the symmetric deformation mode (Eg) and F(3)2g symmetry.40 In addition, a characteristic peak at 160 cm−1 can be observed for LNMO-NRs, which means that the LNMO NRs sample contains trace ordered P4332 phase.41,42 In contrast, the LNMO NPs can be solely indexed to the Fdm space group due to the absence of a splitting of peaks in the 588–623 cm−1 region that is characteristic of ordered spinel structure (P4332).43,44
 | ||
| Fig. 6 Raman spectra of LNMO NPs and NRs. | ||
It is noticed that the degree of cation order in LNMO is closely related to the existence of oxygen vacancies as well as Mn3+. To further confirm this point, XPS experiments of the two LNMO samples were performed and the spectra of the Mn 2p3/2 peaks are presented in Fig. 7. After deconvolution, two kinds of peak positions can be identified, which correspond to the binding energies of Mn3+ and Mn4+ in LNMO similar to those reported in literature.19,40,45 The atomic concentration was further estimated by Gaussian–Lorentz curve fitting and the results indicate a much higher Mn3+ concentration exists in the LNMO NPs than in the NRs. It is thus anticipated the cubic spinel LNMO nanoparticles with disordered Fdm space group and higher Mn3+ concentration would have higher Li+ ion diffusion coefficient and electronic conductivity, as well as better electrochemical performance.
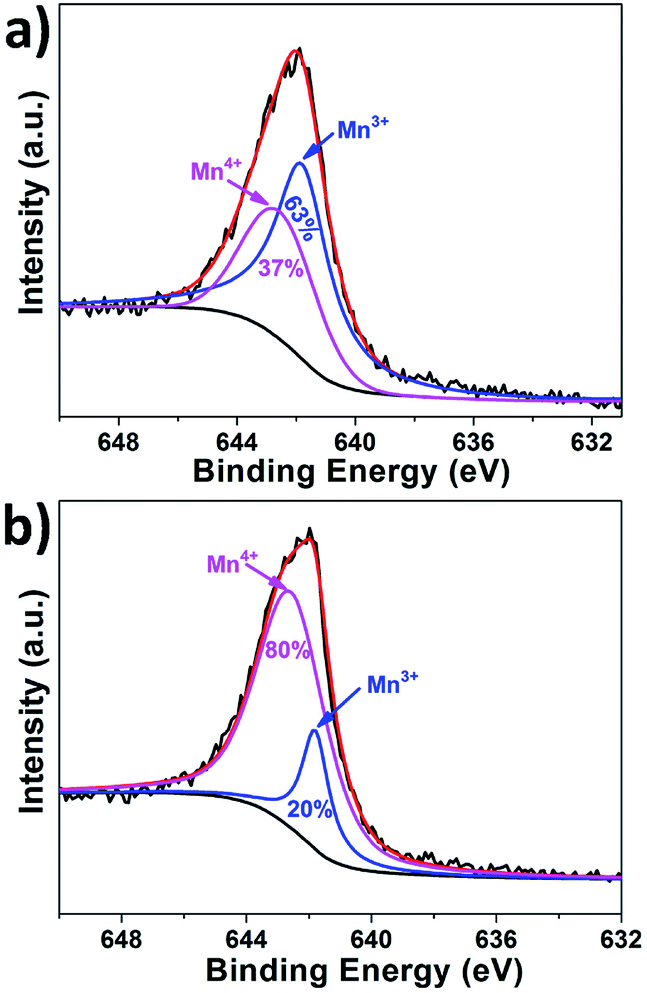 | ||
| Fig. 7 X-ray photoelectron spectra (XPS) of the Mn 2p3/2 peak of (a) LNMO-NPs and (b) LNMO-NRs, respectively. | ||
3.2 The effects of experimental parameters on the synthesis of LNMO NPs
In order to understand the formation mechanism of the spinel LNMO NPs, time-dependent solid-state synthetic experiments were carried out at 750 °C using α-MnO2 nanowires as starting material. Fig. 8 depicts the XRD patterns of the products after calcinations for different time. We can see the LNMO phase was formed after a short reaction period of 1 h (Fig. 8a). Further prolonging calcination time only slightly increases the crystallinity of the LNMO products. However, SEM images indicate significant morphology evolution during the phase transition from tetragonal α-MnO2 to cubic spinel LNMO. In Fig. 9a, the LNMO sample prepared after 1 h consists of nanoparticles with irregular shape and diameters of ∼100 nm on average. After extending the calcination time from 4 to 8 h, the nanoparticles would grow larger and exhibit regular polyhedral shapes (Fig. 9b–d). In addition to the calcination conditions, we find larger and irregular LNMO nanoparticles can be obtained by a similar solid-state reaction using α-MnO2 nanorods as the starting precursor material (Fig. S4†). | ||
| Fig. 8 XRD patterns of the LNMO samples synthesized by solid-state reactions at 750 °C for (a) 1 h, (b) 4 h, (c) 6 h and (d) 8 h, respectively (α-MnO2 nanowires were used as starting material). | ||
 | ||
| Fig. 9 SEM images of the LNMO samples synthesized by solid-state reactions at 750 °C for (a) 1 h, (b) 4 h, (c) 6 h and (d) 8 h, respectively (α-MnO2 nanowires were used as starting material). | ||
Based on the aforementioned results, the formation process of the spinel LNMO nanostructures can be deduced as follows. On the one hand, the MnO2 precursors will experience structural destruction and decompose into other manganese oxides during high temperature calcination process.46,47 On the other, Li+ and Ni2+ ions can migrate into the square channels of the tetragonal MnO2 phase, leading to increased lattice expansion and destabilization of tetragonal structure, as well as formation of new LiO4 tetrahedra and NiO6 octahedra. These synergic effects can accelerate the formation of cubic LNMO nanostructures. As for the α-MnO2 nanowires, their smaller particle diameters and larger 2 × 2 channel can hardly tolerate the structural change and tend to be transformed into more stable LNMO nanoparticles. As for the β-MnO2 nanorods, their larger particle diameters and smaller 1 × 1 pore channels can better tolerate the stress/deformation during the phase transition process, which favours the formation of swollen LNMO nanorods with increased diameters and reduced lengths. In addition, it is noted that the particle size of α-MnO2 also affects the final morphology of LNMO product. In comparison to the α-MnO2 nanorods, the smaller diameters of the α-MnO2 nanowires precursor can facilitate the faster diffusion of Li+ and Ni2+ ions adsorbed on the particle surface and more rapid structure rearrangement, resulting in the formation of well-shaped polyhedral nanoparticles to minimize the total surface energies of the system.
3.3 Electrochemical performances of the LNMO samples
The structure, morphology, particle size and crystal orientation can all affect the electrochemical properties of spinel LNMO materials. In this section, we studied the Li storage capability of LNMO NPs and NRs. Fig. 10a shows the cyclic voltammetry (CV) curves of the LNMO NPs at a scan rate of 0.2 mV s−1 within a potential range of 3.4–5 V (vs. Li+/Li). After the first two cycles, the CV curves in the 3rd and 4th cycles almost overlap, suggesting the full activation and high reversibility of the LNMO NPs electrode. Two pairs of dominant redox peaks located at ∼4.87 V/4.65 V (I1/I2) and ∼4.80 V/4.60 V (II1/II2) can be assigned to the Ni4+/Ni3+ and Ni3+/Ni2+ redox couples, respectively. The peak-to-peak potential separations of the Ni4+/Ni3+ and Ni3+/Ni2+ redox couples are 0.22 V and 0.20 V, respectively. These potential separation values are smaller than or comparable to those reported in the literature,19 suggesting faster lithium ion insertion/extraction kinetics. In addition, another pair of minor redox peaks is noted at around 4.0 V, which stems from the redox reactions of Mn4+/Mn3+ couple (III1/III2) due to the existence of oxygen deficiencies in the disordered spinel LNMO.15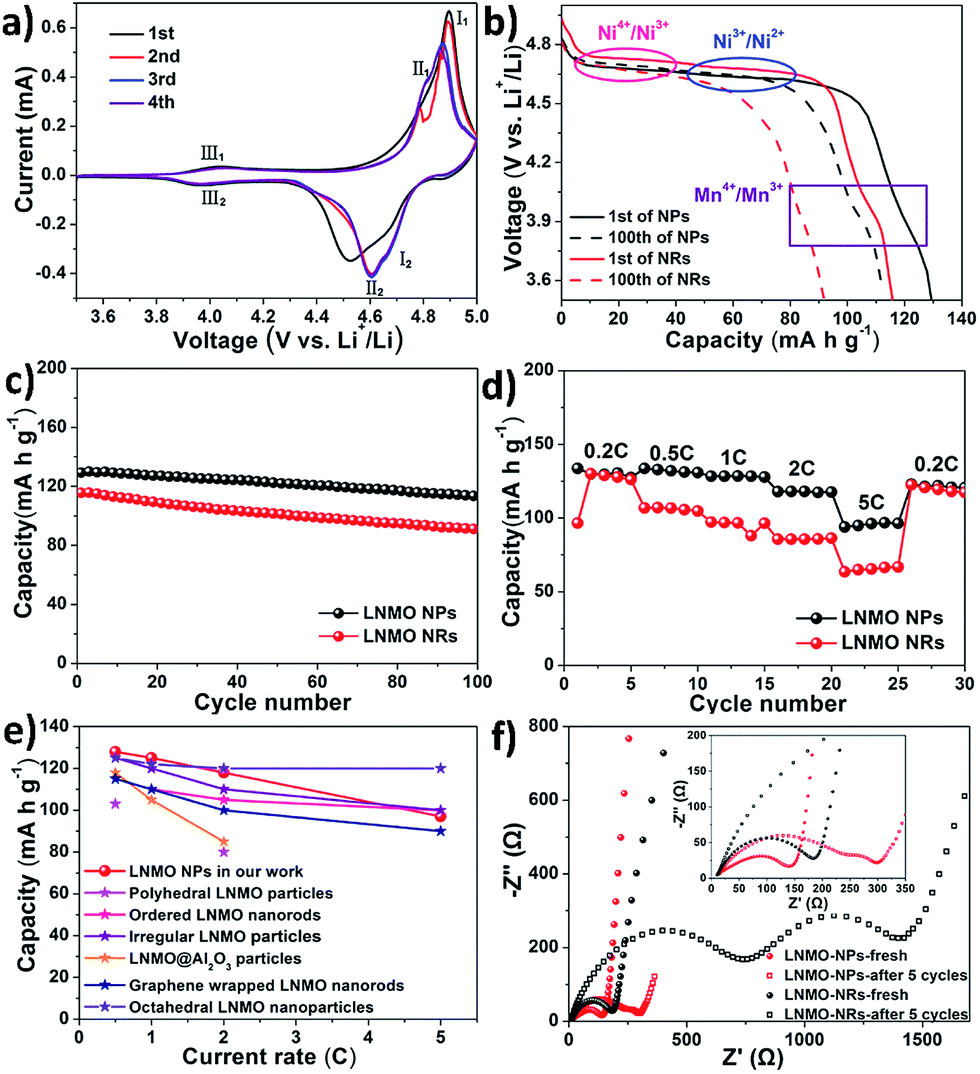 | ||
| Fig. 10 Electrochemical characterizations of the LNMO samples. (a) cyclic voltammetry (CV) curves of LNMO NPs at a scan rate of 0.2 mV s−1; (b) galvanostatic discharge curves of LNMO NPs and NRs at 1C rate (solid line: the 1st cycle, dash line: the 100th cycle); (c) cycling performances of LNMO NPs and NRs subject to galvanostatic test at 1C rate; (d) rate capabilities of LNMO NPs and NRs; (e) comparison of lithium storage capability of LNMO NPs in our work with other LNMO nanostructures in the literature; (f) electrochemical impedance spectra (EIS) of LNMO NPs and NRs (solid: fresh electrode, hollow: after galvanostatic cycling at 0.2C rate for 5 cycles; inset: magnified view of high and high-to-medium frequency regions of EIS spectra). | ||
Fig. 10b and c show the galvanostatic discharge curves and cycling stability of the LNMO NPs and NRs electrodes at 1C rate, respectively. From Fig. 10b, it is evident that the potential plateaus located at ca. 4.7 V in the discharge curves correspond to the electrochemical reduction of Ni4+ ions into Ni3+ ions and Ni2+ ions in sequence, which is in agreement with the CV result shown in Fig. 10a. In addition, a minor discharge plateau located at around 4 V is noted, characteristic of Mn4+/Mn3+ redox couple in disordered LNMO. From Fig. 10c, we can see that both the LNMO NPs and NRs exhibit relatively high Li storage capacity and maintain superior capacity retention during the electrochemical cycling process. In particular, the LNMO NPs manifest a higher initial discharge capacity of ∼129.4 mA h g−1 with a higher capacity retention of 88% over 100 cycles, which is better than those of the LNMO NRs (∼116 mA h g−1 in the 1st cycle with a capacity retention rate of 80% over 100 cycles).
Fig. 10d further compares the rate capabilities of the LNMO NPs and NRs electrodes. Evidently, the LNMO NPs possess higher Li+ ion storage capacities than those of the LNMO NRs when measured at increasing current rates. The discharge capacities of LNMO NPs can be maintained at 127, 130, 127, 117 and 97 mA h g−1 at 0.2, 0.5, 1, 2 and 5C, respectively. After the current rate is lowered to 0.2C again, a discharge capacity of ca. 123 mA h g−1 can be recovered for the LNMO NPs electrode, suggesting its high rate property and excellent cycling stability. In contrast, the discharge capacities of LNMO NRs are 126, 107, 97, 86 and 67 mA h g−1 at 0.2, 0.5, 1, 2 and 5C, respectively, which are inferior to those of the LNMO NPs electrode. In addition, we also notice that the electrochemical performance of our LNMO NPs sample is superior or near to the LNMO nanostructures reported in several literatures, such as polyhedral LNMO particles,48 ordered LNMO nanorods,49 irregular LNMO particles,19 LNMO coated Al2O3 particles,50 graphene wrapped LNMO nanorods,51 and octahedral LNMO nanoparticles.52 A more detailed comparison of electrochemical performance of our LNMO NPs and those in literature is given in Fig. 10e for reference.
Electrochemical impedance spectra (EIS) were further recorded to study the charge transfer kinetics at the electrode–electrolyte interfaces as well as lithium ions diffusion in the solid-state. As shown in Fig. 10f, the Nyquist plots of fresh LNMO electrodes (before electrochemical tests) contain a depressed semicircle at high frequency region and a straight line at low frequency region. The semicircle mainly reflects the charge transfer reaction at electrode–electrolyte interface while the inclined line is an indication of lithium ions diffusion in the solid-state electrode. It can be seen that the diameters of the semicircles for the fresh LNMO NPs and LNMO NRs electrodes are about 150 Ω and 190 Ω, respectively, indicating that the charge transfer occurring at the electrode/electrolyte interface of the former is more facile than the latter. After galvanostatic test at 0.2C for 5 cycles, the EIS spectra comprise two semicircles and one inclined line. The newly emerging semicircle in the high frequency region can be ascribed to the decomposition of moistures adsorbed on the electrode surface as well as the electrolyte decomposition and formation of solid-electrolyte interphase (SEI) layer. Compared to that of LNMO NRs, the lower resistance of the LNMO NPs electrode suggests faster charge transport and transfer at electrode–electrolyte interface.
Ex situ SEM images of the LNMO NPs after galvanostatic test at 1C for 100 cycles are shown in Fig. 11 and S5.† It clearly shows that the morphology of the NPs has been largely preserved during the electrochemical test, proving the high structure stability of the LNMO NPs during electrode preparation and electrochemical measurements.
 | ||
| Fig. 11 SEM image of LNMO NPs after galvanostatic charge–discharge tests at 1C rate for 100 cycles. | ||
The higher Li storage capacity and better rate property of the LNMO NPs can be mainly ascribed to the synergic effect of their distinct structure and geometry characteristics. First, the smaller particle size and relatively narrow particle size distribution of the LNMO NPs can effectively reduce the diffusion lengths of Li ions. Second, the truncated octahedral NPs with dominant {111} facets can maintain a good structural stability with less dissolution of Mn3+ during charge–discharge while the preferential crystal surface orientations of (311), (400), and (440) facets and coarse grain surfaces could facilitate fast Li+ ion insertion/extraction, giving rise to better rate performance.38 Third, high temperature calcinations can produce oxygen deficiency in manganese-based spinel materials and increasing amount of Mn3+ accompanied by an increase in the unit cell volumes.12,53 The higher oxygen vacancy/Mn3+ concentration in the LNMO NPs could improve the electrical conductivity of the spinel cathode and contribute to more storage capacity at ∼4 V region. Also, the larger unit cell volume is beneficial for the Li+ ion insertion/extraction.
4. Conclusions
Well-shaped spinel LiNi0.5Mn1.5O4 nanoparticles have been successfully synthesized by a simple solid-state reaction using α-MnO2 nanowires as the starting materials. The formation of such well-defined polyhedral LNMO nanoparticles was mainly due to the structural destruction of α-MnO2 nanowires during their thermal decomposition followed by preferentially growth of low-energy crystal facets upon high temperature lithiation processes. In contrast, short and bundled LNMO nanorods were obtained when using β-MnO2 nanorods as the starting materials. The well-defined LNMO nanoparticles with truncated octahedral shapes and narrow particle size distribution exhibit high and stable Li storage performance (an initial discharge capacity of 129.4 mA h g−1 at 1C with a capacity retention of 88% over 100 cycles), which are superior to those of the LNMO nanorods (an initial discharge capacity of 116 mA h g−1 and a capacity retention of 80% over 100 cycles). These results indicate that the well-shaped LNMO nanoparticles could be a promising cathode material for high performing Li ion batteries after further structural optimization. The synthesis strategy demonstrated herein is simple and versatile for the potential fabrication of other metal-doped LiMn2O4 and LNMO cathode materials.Acknowledgements
This work is supported by the National Natural Science Foundation of China for Young Scholars (Grant No. 51302204), Shandong Provincial Natural Science Foundation of China (Grant No. ZR2014EMQ011), and Applied Basic Research Foundation of Qingdao City (Grant No. 14-2-4-45-jch). The authors also thank the Research and Test Center of Materials at Wuhan University of Technology for help with TEM and HRTEM analyses as well as Prof. LQ Mai's group for providing electrolyte for lithium-half cells assembly and tests.Notes and references
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Y. Wang, Z.-S. Feng, C. Zhang, L. Yu, J.-J. Chen, J. Hu and X.-Z. Liu, Nanoscale, 2013, 5, 3704–3712 RSC.
- X. Zhang, F. Cheng, J. Yang and J. Chen, Nano Lett., 2013, 13, 2822–2825 CrossRef CAS PubMed.
- Y. Wang, Z.-S. Feng, J.-J. Chen and C. Zhang, Mater. Lett., 2012, 71, 54–56 CrossRef CAS.
- G. Amatucci and J.-M. Tarascon, J. Electrochem. Soc., 2002, 149, K31–K46 CrossRef CAS.
- K. M. Shaju and P. G. Bruce, Chem. Mater., 2008, 20, 5557–5562 CrossRef CAS.
- J.-Y. Luo, H.-M. Xiong and Y.-Y. Xia, J. Phys. Chem. C, 2008, 112, 12051–12057 CAS.
- L. J. Xi, H.-E. Wang, Z. G. Lu, S. L. Yang, R. G. Ma, J. Q. Deng and C. Y. Chung, J. Power Sources, 2012, 198, 251–257 CrossRef CAS.
- Y. L. Ding, J. Xie, G. S. Cao, T. J. Zhu, H. M. Yu and X. B. Zhao, Adv. Funct. Mater., 2011, 21, 348–355 CrossRef CAS.
- R. Santhanam and B. Rambabu, J. Power Sources, 2010, 195, 5442–5451 CrossRef CAS.
- A. Manthiram, K. Chemelewski and E.-S. Lee, Energy Environ. Sci., 2014, 7, 1339–1350 CAS.
- Q. Zhong, A. Bonakdarpour, M. Zhang, Y. Gao and J. Dahn, J. Electrochem. Soc., 1997, 144, 205–213 CrossRef CAS.
- J.-H. Kim, S.-T. Myung, C. Yoon, S. Kang and Y.-K. Sun, Chem. Mater., 2004, 16, 906–914 CrossRef CAS.
- S. Park, S.-W. Oh, S. Kang, I. Belharouak, K. Amine and Y.-K. Sun, Electrochim. Acta, 2007, 52, 7226–7230 CrossRef CAS.
- K. R. Chemelewski, E.-S. Lee, W. Li and A. Manthiram, Chem. Mater., 2013, 25, 2890–2897 CrossRef CAS.
- M. Kunduraci and G. Amatucci, J. Electrochem. Soc., 2006, 153, A1345–A1352 CrossRef CAS.
- L. Wang, H. Li, X. Huang and E. Baudrin, Solid State Ionics, 2011, 193, 32–38 CrossRef CAS.
- Y. Xue, Z. Wang, F. Yu, Y. Zhang and G. Yin, J. Mater. Chem. A, 2014, 2, 4185–4191 CAS.
- C. J. Jafta, M. K. Mathe, N. Manyala, W. D. Roos and K. I. Ozoemena, ACS Appl. Mater. Interfaces, 2013, 5, 7592–7598 CAS.
- M.-H. Liu, H.-T. Huang, C.-M. Lin, J.-M. Chen and S.-C. Liao, Electrochim. Acta, 2014, 120, 133–139 CrossRef CAS.
- T.-F. Yi, B. Chen, Y.-R. Zhu, X.-Y. Li and R.-S. Zhu, J. Power Sources, 2014, 247, 778–785 CrossRef CAS.
- T.-F. Yi and X.-G. Hu, J. Power Sources, 2007, 167, 185–191 CrossRef CAS.
- Y. Lin, Y. Yang, R. Yu, H. Lai and Z. Huang, J. Power Sources, 2014, 259, 188–194 CrossRef CAS.
- S.-Y. Bae, W.-K. Shin and D.-W. Kim, Electrochim. Acta, 2014, 125, 497–502 CrossRef CAS.
- H. Lin, Y. Zhang, J. Hu, Y. Wang, L. Xing, M. Xu, X. Li and W. Li, J. Power Sources, 2014, 257, 37–44 CrossRef CAS.
- W. Wu, H. Xiang, G. Zhong, W. Su, W. Tang, Y. Zhang, Y. Yu and C. Chen, Electrochim. Acta, 2014, 119, 206–213 CrossRef CAS.
- Y. Liu, M. Zhang, Y. Xia, B. Qiu, Z. Liu and X. Li, J. Power Sources, 2014, 256, 66–71 CrossRef CAS.
- D. K. Kim, P. Muralidharan, H.-W. Lee, R. Ruffo, Y. Yang, C. K. Chan, H. Peng, R. A. Huggins and Y. Cui, Nano Lett., 2008, 8, 3948–3952 CrossRef CAS PubMed.
- E. Hosono, T. Kudo, I. Honma, H. Matsuda and H. Zhou, Nano Lett., 2009, 9, 1045–1051 CrossRef CAS PubMed.
- H.-W. Lee, P. Muralidharan, R. Ruffo, C. M. Mari, Y. Cui and D. K. Kim, Nano Lett., 2010, 10, 3852–3856 CrossRef CAS PubMed.
- X. Wang and Y. Li, J. Am. Chem. Soc., 2002, 124, 2880–2881 CrossRef CAS PubMed.
- X. Su, X. Yang, L. Yu, G. Cheng, H. Zhang, T. Lin and F.-H. Zhao, CrystEngComm, 2015, 17, 5970–5977 RSC.
- H.-E. Wang, D. Qian, Z. Lu, Y. Li, R. Cheng and Y. Li, J. Phys. Chem. Solids, 2007, 68, 1422–1427 CrossRef CAS.
- J. Xiao, P. Liu, Y. Liang, H. Li and G. Yang, J. Appl. Phys., 2013, 114, 073513 CrossRef.
- Z. Li, Y. Ding, Y. Xiong, Q. Yang and Y. Xie, Chem. Commun., 2005, 918–920 Search PubMed.
- H.-E. Wang, Z. Lu, D. Qian, S. Fang and J. Zhang, J. Alloys Compd., 2008, 466, 250–257 CrossRef CAS.
- H. Wang, Z. Lu, D. Qian, Y. Li and W. Zhang, Nanotechnology, 2007, 18, 115616 CrossRef.
- J.-S. Kim, K. Kim, W. Cho, W. H. Shin, R. Kanno and J. W. Choi, Nano Lett., 2012, 12, 6358–6365 CrossRef CAS PubMed.
- C. Julien and M. Massot, Mater. Sci. Eng., B, 2003, 97, 217–230 CrossRef.
- Y. Wei, L. Yan, C. Wang, X. Xu, F. Wu and G. Chen, J. Phys. Chem. B, 2004, 108, 18547–18551 CrossRef CAS.
- J. Song, D. W. Shin, Y. Lu, C. D. Amos, A. Manthiram and J. B. Goodenough, Chem. Mater., 2012, 24, 3101–3109 CrossRef CAS.
- X. Zhu, X. Li, Y. Zhu, S. Jin, Y. Wang and Y. Qian, Electrochim. Acta, 2014, 121, 253–257 CrossRef CAS.
- X. Zhang, F. Cheng, K. Zhang, Y. Liang, S. Yang, J. Liang and J. Chen, RSC Adv., 2012, 2, 5669–5675 RSC.
- N. Amdouni, K. Zaghib, F. Gendron, A. Mauger and C. Julien, Ionics, 2006, 12, 117–126 CrossRef CAS.
- K. Shaju, G. S. Rao and B. Chowdari, Electrochim. Acta, 2003, 48, 1505–1514 CrossRef CAS.
- B. Gates, Y. Wu, Y. Yin, P. Yang and Y. Xia, J. Am. Chem. Soc., 2001, 123, 11500–11501 CrossRef CAS PubMed.
- B. Gates, B. Mayers, Y. Wu, Y. Sun, B. Cattle, P. Yang and Y. Xia, Adv. Funct. Mater., 2002, 12, 679–686 CrossRef CAS.
- A. Bhaskar, N. N. Bramnik, A. Senyshyn, H. Fuess and H. Ehrenberg, J. Electrochem. Soc., 2010, 157, A689–A695 CrossRef CAS.
- H.-W. Lee, P. Muralidharan, C. M. Mari, R. Ruffo and D. K. Kim, J. Power Sources, 2011, 196, 10712–10716 CrossRef CAS.
- J. W. Kim, D. H. Kim, D. Y. Oh, H. Lee, J. H. Kim, J. H. Lee and Y. S. Jung, J. Power Sources, 2015, 274, 1254–1262 CrossRef CAS.
- X. Tang, S. S. Jan, Y. Qian, H. Xia, J. Ni, S. V. Savilov and S. M. Aldoshin, Sci. Rep., 2015, 5, 11958 CrossRef PubMed.
- H. Lin, Y. Zhang, H. Rong, S. Mai, J. Hu, Y. Liao, L. Xing, M. Xu, X. Li and W. Li, J. Mater. Chem. A, 2014, 2, 11987–11995 CAS.
- D. Pasero, N. Reeves, V. Pralong and A. West, J. Electrochem. Soc., 2008, 155, A282–A291 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21723g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |