
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu(I) N-heterocyclic carbenes from bis-imidazolium amphiphiles: synthesis, cytotoxicity and incorporation onto gold nanoparticles†
M.
Rodrigues
 a,
L.
Russo
a,
E.
Aguiló
b,
L.
Rodríguez
b,
I.
Ott
c and
L.
Pérez-García‡
*a
a,
L.
Russo
a,
E.
Aguiló
b,
L.
Rodríguez
b,
I.
Ott
c and
L.
Pérez-García‡
*a
aDepartament de Farmacologia i Química Terapèutica and Institut de Nanociència i Nanotecnología UB (IN2UB), Universitat de Barcelona, Avda. Joan XXIII s/n, 08028 Barcelona, Spain. E-mail: mlperez@ub.edu
bDepartament de Química Inorgànica, Universitat de Barcelona, C/Martí i Franquès 1-11, 08028 Barcelona, Spain
cInstitute of Medicinal and Pharmaceutical Chemistry, Technische Universität Braunschweig, Beethovenstr. 55, 38106 Braunschweig, Germany
First published on 22nd December 2015
Abstract
A gold(I) N-heterocyclic carbene 4 from a bis-imidazolium-amphiphile was synthesized and characterized. The cytotoxicity against HT-29 colon carcinoma and MDA-MB-231 breast adenocarcinoma cells was assessed for the NHC complex 4, the imidazolium salt precursor 2, and its methyl analogue 3, indicating that compounds 2–4 are promising cytotoxic agents. Furthermore, the ability of these compounds to be associated with gold nanoparticles was also explored, in order to develop an anticancer drug delivery system. The free ligands displayed more activity when compared with the ligands immobilized on the gold nanoparticles. The synthesized gold particles incorporating the bis-imidazolium salts either 2 or 3 showed monodisperse spherical shape with sizes of approximately 5 nm.
Introduction
One of the most urgent and challenging objectives of medicinal chemistry is the discovery of novel and more effective drugs for fighting cancer. Bioinorganic and bio-organometallic chemistry provides different examples of metal complex-based drugs whose tumour activity is well known: among them, cisplatin1 is one of the most frequently used clinical anticancer drugs. The gold compound auranofin2,3 represents an important milestone in the development of anticancer drugs based on Au(I)–phosphine complexes.4 Other phosphine–Au(I) derivatives with anticancer activity include phosphine–Au(I)–thiolates, phosphine–Au(I)–phosphines, phosphine–Au(I)-alkynyls or phosphine–Au(I)-halides.4 Some phosphine–Au(I)-alkynyl derivatives present higher activity than cisplatin in human ovarian cancer cells5 and on Hep3B hepatocellular carcinoma cells.6Thioredoxin reductase (TrxR) is an important and ubiquitous enzyme critically involved in the regulation of intracellular metabolism. The thioredoxin/thioredoxin reductase system plays an important role in the redox state of the cells, besides being involved in other cell functions such as cell proliferation, transcription factor regulation or apoptosis.7 Inhibition of this system causes oxidized thioredoxin to accumulate in cells, promoting apoptosis, thus making thioredoxin reductase a good target for cancer therapy.8,9 In this context, increasing interest has grown towards the development of inhibitors based on lipophilic Au(I) complexes.10 These complexes are a very promising class of non-platinum based antitumor agents, and among them neutral and cationic complexes with N-heterocyclic carbenes (NHCs) as ligands show a remarkable strong anti-cancer effect through the inhibition of Trx/TrxR reductase system.10 Recent results indicate that TrxR inhibition in combination with antimitochondrial effects are key properties that determine the bioactivity of gold NHC complexes. This therapeutic strategy exploits the negative mitochondrial membrane potentials to selectively concentrate delocalised lipophilic cations, such as Au(I) complexes, within the organelle. In addition, the gold complex provides the compound the ability to selectively coordinate the enzyme's functional selenocysteine, increasing its anti-tumour activity.10
N-Heterocyclic carbenes (NHC) metal complexes11,12 are known and studied as potential anticancer metallodrugs because, compared with traditional gold complexes such as auranofin, they show an enhanced stability of the coordinative bond with the metal atom and therefore they likely do not undergo fast metabolization before reaching their enzymatic target.13 Despite the similarities with the well-known phosphine–gold complexes,4 NHC ligands represent a better alternative due to their stability, as well as the ease with which it can be functionalized in order to vary their lipophilicity. Nowadays, different groups concentrate their investigation on developing novel gold NHC complexes with anti-cancer properties. Schuh et al. successfully synthesized a family of substituted imidazole and benzimidazole asymmetric Au(I) linear NHCs, evaluating both their antiproliferative effects on human ovarian cancer cell lines and their effective inhibition of TrxR.14 Within the same type of NHC–Au–L compounds, Rubbiani et al. compared the effect of different ligand L (L = –Cl, –NHC, or –PPh3) on TrxR inhibition and antimitochondrial action,15 showing how both the cationic character and the modulation of the stability of the coordinative bond of the complexes are key features for their cytotoxicity. Liu et al. also provides a further example of a diarylimidazole-based gold NHCs, in which a correlation between complexes' biological activity and both the inactive, non-NHC ligand and the aromatic substituents can be established.16 We have also described the synthesis of Au(I) N-heterocyclic monocarbenes and dicarbenes derived from imidazolium salts, their biological activity and TrxR inhibition ability.17
Moreover, recent works provide evidence of the wide range of functionalities that can be introduced in this class of heterocyclic compounds in order to further investigate their antitumor activity: Pratesi et al.18 explored the inhibition mechanism of TrxR by successfully coupling three different Au(I)–NHC complexes with a synthetic dodecapeptide containing a selenocysteine group, confirming the direct coordination of this thiol-residue with the gold atom. Citta et al. took advantage of the ease of modification of NHCs for providing their complexes with a fluorescent anthracene unit. This functionalization not only does not affect the gold compound's antiproliferative effect, but also allows the study of its distribution in vivo.19
In our group, a class of gemini-type imidazolium surfactants has been extensively studied because of its anion recognition properties20,21 as well as its capability of stabilizing gold nanoparticles (AuNPs) synthesized in a biphasic system.20,22 The amphiphilic bis-imidazolium compounds play a double role in the synthesis, acting as transfer agents and stabilizers. Furthermore, their anion recognition ability allowed the AuNPs to incorporate successfully a model anionic drug and release it in a sustained manner. Additionally, by changing the synthetic method, we were able to produce amphiphiles bilayer-coated water-soluble AuNPs that could load and deliver piroxicam, an anti-inflammatory drug with poor water solubility.23 Therefore, these imidazolium-based molecules are promising materials for biomedical applications.24 It would be interesting to go one step further and add a novel role to these molecules by introducing a metal complex with gold moiety, thus achieving AuNPs that would have not only the ability to carry drugs, but also present biological activity due to the presence of the N-heterocyclic carbenes (NHC) metal complex. The strategy to achieve this goal would include synthesizing the NHC–Au(I) complex, but also to take advantage of the versatility of these molecules, a propargyloxy group was introduced in order to allow the formation of a phosphine–Au(I)-alkynyl derivative.
Thus, in this study we develop the synthesis of novel bis-imidazolium Au(I) carbene complexes as multifunctional ligands for the synthesis of AuNPs with potential anticancer activity, and we report: (a) the synthesis and structural characterization of a bis-imidazolium gold(I) complex, (b) the synthesis and morphological characterization of AuNPs, bearing different kinds of bis-imidazolium amphiphilic ligands as stabilizing agents, and (c) the evaluation of the biological activity of both the ligands and AuNPs.
Results and discussion
Synthesis of Au(I) complexes
The reaction of DBMPB25 with N-octadecylimidazole (1a)20 or 2-methyl-N-octadecylimidazole (1b) following a similar protocol allowed obtaining the two bis-imidazolium amphiphiles 2 and 3 respectively (Scheme 1) in almost quantitative yields.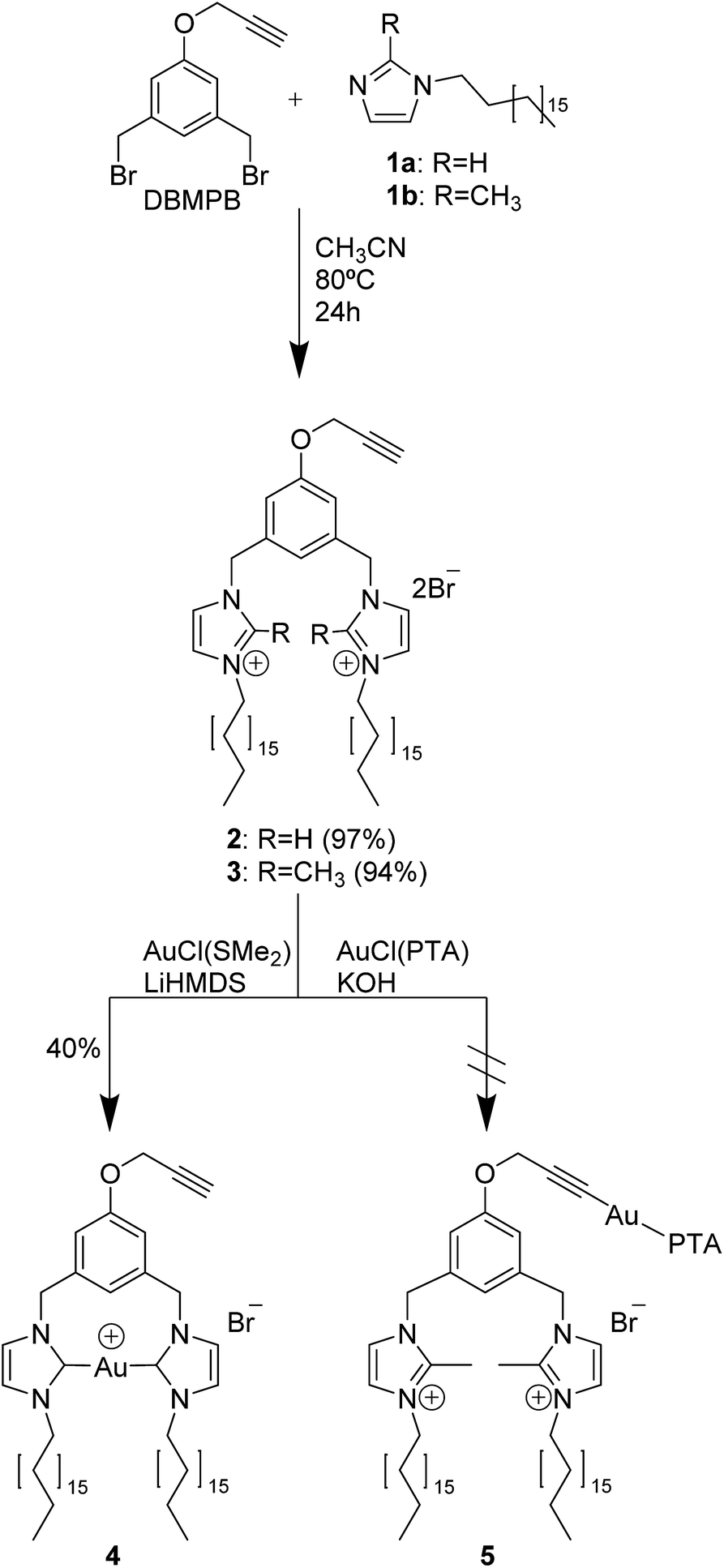 | ||
| Scheme 1 Synthesis of imidazolium-based compounds 2 and 3, formation of the carbene complex 4 and attempt of the formation of complex 5 with phosphine–Au(I)-alkynyl derivative. | ||
Two parallel strategies were then followed in order to obtain the imidazolin-2-ylidene-Au(I) complex 4 (Scheme 1). The aim was to direct selectively the ligand substitution reaction of the gold precursor towards one precise functional group by varying the reaction conditions and reagents. In the first strategy, by following a general procedure described in literature,26 we targeted the acidic proton of the imidazolium rings, in order to form a neutral heterocyclic carbene (NHC) as a coordinating reactive site. Compound 4 was successfully synthesized by the reaction of [AuCl(SMe2)] with 2, deprotonated in situ with lithium bis(trimethylsilyl)amide (LiHMDS) in anhydrous acetonitrile.11 This procedure provided selective conditions for the synthesis of the Au(I)-carbene complex 4, obtained as a colourless solid by recrystallization from acetone–hexane or ethanol–diethylether with a 40% yield. The second strategy focused on targeting the terminal alkyne of the propargylic ether by deprotonating it in situ with potassium hydroxide, promoting the coordination with a gold-1,3,5-triaza-7-phosphaadamantane-chloride complex [AuCl(PTA)].27 Despite the relatively mild basic environment, deprotonation was not obtained selectively on the propargylic ether of compound 2 but a competition with the imidazolium acidic proton was observed. Various obstacles during purification were encountered, probably due to the lack of regioselectivity, thus an univocal structure was not identified. To solve this inconvenience, the same reaction was carried out on the methylated bis-imidazolium ligand 3, in an attempt to obtain the complex 5 incorporating a phosphine–Au(I)–alkyinyl derivative. However due to the tendency towards aggregation caused by the bulky PTA ligand, the desired gold complex could not be isolated in its pure form and consequently was not successfully characterized.
Compounds 2, 3 and 4 were characterized by 1H NMR spectroscopy. The corresponding spectroscopy data is summarized in Table 1.

While both the imidazolium dications maintain similar chemical shifts for their analogous groups (for instance the terminal alkyne proton peak at 2.64 ppm and 2.57 ppm respectively), the imidazolium substituent R in position 2 was indicative for distinguishing between the two compounds. The formation of Au(I) carbene complex 4 was also determined by 1H NMR spectroscopy (Table 1) by following the disappearance of the imidazolium acidic proton signal at 10.44 ppm, which might indicate the coordinative bond formation between the imidazolium dicarbene and the gold atom. The regioselectivity of the reaction was proved since Au(I) compound spectra presented always the peak corresponding to the alkynyl terminal proton at 3.53 ppm, with a Δδ = +0.89 ppm compared to the starting material.
The structures of compounds 2 and 3 were also confirmed by Electrospray Ionization Mass Spectrometry (ESI-MS) with both presenting a main characteristic fragmentation ion corresponding to the dication resulting of the loss of the two bromide counterions [(M − 2Br)/2]2+ (Table 2 and ESI, Fig. S1 and S2†).
| MWb (g mol−1) | Ionsa (m/z) | |||
|---|---|---|---|---|
| [(M − 2Br)/2]2+ | [M − Br]+ | [M–Br–CH2CCH−]+ | ||
| a Ions, m/z ratio relative abundance (%). b Molecular weight. c ESI-MS. d MALDI-TOF-MS with matrix DHB. e MALDI-TOF-MS without matrix. | ||||
| 2 c | 959.1 | 399.4 (100%) | ||
| 3 c | 987.2 | 413.4 (100%) | ||
| 4 d | 1074.2 | 993.8 (100%) | 955.7 (25%) | |
| 4 e | 993.7 (75%) | 955.7 (100%) | ||
Further confirmation of Au(I) carbene 4 was provided through Matrix Assisted Laser Desorption Ionization Time-Of-Flight Mass Spectrometry (MALDI-TOF MS) analysis which showed, in the case of DHB-supported ionization, two main peaks, the most abundant peak corresponding to bromide loss (M − Br)+ at 993.80 m/z, in accordance with previously described analogues,20 and the second peak from the simultaneous bromide and propargyl loss (M − Br − CH2CCH)+ at 955.70 m/z (see ESI, Fig. S3†). The measurement was also performed without polymer matrix and the same main characteristic signals were observed, together with peaks related to the gradual fragmentation of the alkyl chains (see ESI, Fig. S4†). There were however some differences observed in the abundance of the obtained peaks: when the analysis was performed without matrix, the most abundant peak corresponded to the loss of bromide and propargyl, whereas in the case of the analysis performed with matrix, the abundance is much lower (25%), and the peak corresponding to the molecule with no loss of bromide is also identified with low abundance (10%).
Synthesis of gold nanoparticles (AuNPs)
Our group reported that bis-imidazolium derived gemini-type surfactants are able to recognize anions,20–22 an interesting feature that can be used in nanomedical applications, and described how these molecules could also be used for the synthesis and stabilization of AuNPs through a modification of the Brust–Schiffrin method.20,22 The synthesis of AuNPs is achieved through reduction of HAuCl4 with NaBH4 in a biphasic system, in which the imidazolium salts carry out both the functions of phase transfer agent and stabilizer, besides maintaining the ability to recognize and complexate anions, making them a promising tool for delivery applications together with gold nanoparticles as drug carriers.Following the previously reported procedure, the nanoparticle synthesis was initially carried out successfully using the free bis-imidazolium ligands 2 and 3 through the modified Brust–Schiffrin method. The AuNPs formation was monitored by UV-visible absorption spectroscopy, following the characteristic Surface Plasmon Resonance (SPR) band at ca. 520 nm (Fig. 1). The samples were further identified as 2·AuNP and 3·AuNP, respectively. After washing, the absorption spectrum was compared to the free ligand, whose peaks at around λ = 280 nm are clearly recognizable in the nanoparticles solution, confirming its presence as stabilizer.
 | ||
| Fig. 1 UV-visible absorption spectra of free ligand 2 (grey) and 2·AuNP in CH2Cl2 (black). The peaks corresponding to the SPR band (523 nm) and the imidazolium ligand (280 nm) are visible in the 2·AuNP spectrum. | ||
Additionally, compound 2 and the corresponding 2·AuNP sample were analysed by Infrared spectroscopy (IR) and it was possible to identify the peaks of the ligand 2 on the AuNPs (see ESI, Fig. S5 and S6†). The peaks corresponding to the C–H bond in the imidazolium ring can be found around 3100 cm−1 (being less visible in the 2·AuNP spectrum). Two pronounced peaks, corresponding to the CH2 from the alkyl chains are found at 2850 and 2920 cm−1, around 2100 cm−1 a peak assigned to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C is found (also less pronounced in the 2·AuNP spectrum). Between 1560 and 1650 cm−1 three peaks, that can be assigned to the C
C is found (also less pronounced in the 2·AuNP spectrum). Between 1560 and 1650 cm−1 three peaks, that can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN from the imidazolium moieties, are found. Generally, the peaks of imidazolium salts are less intense, especially the ones from the functional groups which are somehow interacting with the metallic gold, such as the phenyl ring and the alkine.
C and CN from the imidazolium moieties, are found. Generally, the peaks of imidazolium salts are less intense, especially the ones from the functional groups which are somehow interacting with the metallic gold, such as the phenyl ring and the alkine.
The synthesized AuNPs were observed by High Resolution Transmission Electron Microscopy (HRTEM) to study their morphology and size. The obtained micrographs can be seen in Fig. 2 and the size distribution can be observed in the corresponding included histograms.
 | ||
Fig. 2 HRTEM micrographs of 2·AuNP with (a) 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000x and (b) 100000x, magnification, and of 3·AuNP with (c) 25000x and (d) 150000x magnification, and their corresponding histograms. 000x and (b) 100000x, magnification, and of 3·AuNP with (c) 25000x and (d) 150000x magnification, and their corresponding histograms. | ||
It is possible to identify relatively monodisperse spherical particles with average diameter of 4.8 ± 1.1 nm and 4.2 ± 1.0 nm for 2·AuNP and 3·AuNP respectively.
By comparing the 2·AuNP and 3·AuNP nanoparticles, formed with either imidazolium ligand 2 or 3, both TEM images and UV-vis absorption spectroscopy did not provide any evidence of relevant morphological difference, suggesting that the introduction of a methyl chain in the imidazolium ring did not affect significantly the ability of the ligand 3 to stabilize AuNPs.
X-ray photoelectron spectroscopy (XPS) showed the presence of two peaks at 84.2 eV and 87.8 eV (in the case of 2·AuNP) and 85.2 eV and 88.8 eV (in the case of 3·AuNP) corresponding to Au4f7/2 and Au4f5/2 respectively. The peak position and the distance between the two peaks (3.7 eV) is consistent with gold in its reduced form Au(0) (see ESI, Fig. S7†) and is in agreement with previously described bis-imidazolium coated nanoparticles.20 Furthermore, the presence of minor peaks corresponding to Au(I), that can be found with a shift of 1–2 eV with respect to the Au(0) peaks,28–30 were not identified during the curve fit of the XPS spectra, suggesting that no carbene species were formed in situ during the AuNP synthesis, and that the interaction between the ligand and the gold surface is not mediated by carbenes.
The AuNPs were also analysed by X-ray diffraction (XRD) to determine the phase composition. Four Bragg peaks could be identified in the gold XRD pattern, indicating a face-centered cubic (fcc) structure, with peaks at 37°, 44°, 65° and 78°, corresponding respectively to (111), (200), (220) and (311) planes (see ESI, Fig. S8†).
Thermogravimetric analysis was performed on 2·AuNP. The data obtained for the amount of ligand present in the sample, together with size data obtained from size measurements, allowed the calculation of the number of ligands present in the AuNPs (see ESI, Fig. S9 and Table S1†). It was possible to determine that the AuNPs sample has approximately 24 ligands per nm2. This value is in agreement with previous described AuNPs with similar ligands, that present around 28–30 ligands per nm2.22,23 This data also allows to determine the amount of ligands present per gold nanoparticle, thus allowing to calculate the approximate molecular weight of the whole nanostructure.
Following these results, experiments were made to synthesize the Au(I)-complex directly on the formed 2·AuNP nanoparticles, with compound 2 as stabilizing agent. The goal was to obtain the advantages of the gold nanoparticle as drug carrier, and at the same time have a ligand with anticancer activity. To perform the synthesis of the NHC metal complex, the same procedure described in Scheme 1 was attempted, but the basic medium caused the colloid to precipitate, and this approach was discarded.
Cytotoxicity study
The biological activity for the gold NHC 4 and for the bis-imidazolium salt 2 and its methyl analogue 3 alone or conjugated with AuNP (2·AuNP and 3·AuNP) was evaluated. Therefore, the cytotoxicity against tumour cell growth was determined using the cell lines HT-29 (colon adenocarcinoma) and MDA-MB-231 (breast adenocarcinoma). The antiproliferative activity was expressed as IC50 values for cytotoxicity in both cell lines, and the values are presented in Table 3.From the above values it can be observed that the free compounds do not follow a trend in the two studied cell lines as the observed IC50 values are within a rather narrow range (6–13 μM). For complex 4, due to limited solubility this compound could not be administered at concentrations higher than 10 μM, and it was found that the IC50 against HT-29 cells is higher than the maximum concentration tested.
The nanoparticles 2·AuNP and 3·AuNP were both applied at maximum dosages of 0.0044 μM and did not display cytotoxicity at this concentration. Using the data from the TGA analysis, it is possible to determine the molecular weight of the AuNP and to determine the amount of ligand present per mole of AuNP. The calculations made show that to the applied concentration of 0.0044 μM of AuNP corresponds a concentration of 16 μM of ligand. This value is similar to the IC50 found for the free compounds, which means that when bound to the AuNPs, the ligands do not present the same toxic effect. However, since this was the maximum concentration tested, it was not possible to determine precisely the IC50 value of the ligand bound to the AuNPs.
These findings are in agreement with the results obtained with a previously described analogue,20 also studied free and conjugated to AuNPs, where we found a similar outcome. The IC50 value (determined in Caco-2 cells) is similar to the one found for these compounds, but the corresponding AuNPs cytotoxicity was also higher than the maximum tested concentration. Therefore, these results suggest that the ligands alone present higher cytotoxicity, because they are more available for interaction with the cells.
Conclusions
In this work we describe the synthesis of two bis-imidazolium precursors and a derived carbene complex, all showing cytotoxic effect on two different adenocarcinoma cell lines. Furthermore, the bis-imidazolium complexes could be used successfully to synthesize gold nanoparticles. However, when incorporated on the AuNP no cytotoxicity was observed. This behaviour was explained by the fact that the positive charges from the polar head are not available to interact with the cells because they are involved in the stabilization of the nanoparticles and hidden in the lipophilic layer. Further work includes the formation of phosphine–Au(I)-alkynyl derivatives with the alkyne moiety of ligands 2 and 3 after the formation of the gold nanoparticles, and also the formation of gold nanoparticles with the carbene complex 4.Experimental section
Materials
All solvents used were of analytical grade. Acetonitrile was dried over basic aluminium oxide. Anhydrous tetrahydrofuran was obtained through distillation over Na/benzophenone. Commercial compounds: 1-bromooctadecane, chloro(dimethyl sulfide)gold(I), hydrogen tetrachloroaurate (III), lithium bis(trimethylsilyl)amide, 1,3,5-triaza-7-phosphaadamantane (PTA) and sodium borohydride were purchased from Aldrich. 2-Methylimidazole was purchased from Fluka. Potassium hydroxide (KOH pellets) 85% was purchased from Scharlau. CDCl3 was purchased from Euriso Top. Methods described in literature were used to prepare 1,3-bisbromoethyl-5-propargyloxybenzene (DBMPB)25 and N-octadecylimidazole.20General methods
1H NMR and 13C NMR were performed on Varian Gemini 300 and Varian Mercury 400 spectrometers from the Centres Científics i Tecnològics de la Universitat de Barcelona (CCiT-UB). NMR spectra were determined in CDCl3 and chemical shifts are expressed in parts per million (δ) relative to TMS. Thin layer chromatography was performed on Merck coated 60 F254 silica gel plates; the spots were located with UV light and developed with an iodine/silica or 1% (w/v) potassium permanganate solution. UV-visible absorption spectra were determined in CH2Cl2 on a UV-1800 Shimadzu UV spectrophotometer. ESI-MS was performed on a LC/MSD-TOF mass spectrometer from Agilent Technologies: the nebulizing nitrogen gas flow at 15 psi and drying nitrogen 7 L min−1; the source temperature was maintained at 300 °C with a capillary voltage of 4 kV and a fragment or voltage of 80 or 215 V. The eluent flowing through the probe was H2O:CH3CN (1:1) at a flow rate of 200 μL min−1. High resolution mass spectra (HR-MS): electro-spray (ESI) with high resolution in Agilent (2006) LC/MSD-TOF mass spectrometer. MALDI-TOF-MS were performed on a 4800 Plus MALDI TOF/TOF mass spectrometer from AB Sciex, from CCiT-UB. The laser operated at 355 nm, at a frequency of 200 Hz and at a pulse of 3–7 ns. DHB (2,5-dihydroxybenzoic acid) was used as matrix. HRTEM images were obtained using a JEOL JEM 2100 transmission electron microscope at 200 kV, from CCiTUB. The images were captured by a Megaview III Soft Imaging System camera, and the size of the nanoparticles gold core was measured with ImageJ. TGA was performed on a Mettler Toledo TGA/SDTA 851e, at CCiTUB, with a temperature ramp from 30 °C to 600 °C and a heating rate of 10 °C min−1. XPS analysis was carried out at CCiT-UB using a Kratos Axis Ultra spectrometer with monochromatic Al Kα (1486.6 eV, 10 mA, 15 kV) X-ray source and a power of 150 W. Pressures near 1 × 10−8 Torr were observed in the analytical chamber during surface analysis. All XPS analysis employed an analysis area of 700 μm × 300 μm. Survey spectra were recorded using analyzer pass energy of 160 eV and a 1.0 eV energy step. XRD measurements were performed with a Siemens D-5000 X-ray diffractometer from the Scientific Experimental Platforms, ICMAB-CSIC. The source was a DRX ceramic tube (λ CuKα = 1.540560 Å and λ CuKα2 = 1.544390 Å) with a voltage and current of 45 kV and 35 mA, respectively. The AuNP samples were placed on a glass slide and dried under vacuum, and were scanned from 2θ = 30–80°.
Synthesis of compounds
1b . 1H NMR (300 MHz, CDCl3): 0.88 (t, J = 6.8 Hz, 3H, –CH3); 1.25 (m, 30H, –(CH2)15–); 1.73 (m, 2H, –NCH2CH2–); 2.37 (s, 3H, Im–CH3); 3.80 (t, J = 7.2 Hz, 2H, –NCH2–); 6.80 (d, J = 1.5, 1H, Im–H); 6.90 (d, J = 1.2, 1H, Im–H).
2 . 1H NMR (300 MHz, CDCl3): 0.86 (t, J = 6.3 Hz, 6H, –CH3); 1.26–1.32 (m, 60H, –(CH2)15–); 1.88 (m, 4H, –NCH2CH2–); 2.64 (t, J = 2.3 Hz, 1H, –CCH); 4.24 (t, J = 7.5 Hz, 4H, –NCH2–); 4.45 (d, J = 2.3 Hz, 2H, –OCH2–); 5.57 (s, 4H, –CH2–); 7.20 (s, 2H, Im–H); 7.26 (s, 2H, Im–H); 7.76 (s, 1H, Ar-H); 8.33 (s, 2H, Ar-H); 10.44 (s, 2H, Im–H). 13C (100 MHz, CDCl3): 14.3 (–CH2(CH2)13CH3); 32.1–22.8 (–CH2(CH2)13CH3); 50.4 (–CH2(CH2)13CH3); 52.5 (–Ar-CH2N–); 56.7 (–CH2–CCH); 76.8 (–CH2CCH); 116.5 (Im1); 121.6 (Im2); 123.7 (Ar2); 124.1 (Ar1); 136.2 (–ArCCH2N–); 137.0 (Im–H); 158.3 (–ArCO–). IR: 2111 cm−1 (∂ C–C, terminal alkyne). MS (ESI) (m/z, %): 399.4, [(M − 2Br)/2]2+, 100%. Rf: 0.5 (DCM
:MeOH 95:5). mp: 160 °C.
3 . 1H NMR (300 MHz, CDCl3): 0.88 (t, J = 6.6 Hz, 6H, –CH3); 1.26–1.32 (m, 60H, –(CH2)15–); 1.80 (m, 4H, –NCH2CH2–); 2.57 (s, 1H, –CCH); 2.78 (s, 6H, Im–CH3); 4.17 (m, 4H, –NCH2–); 4.79 (m, 2H, –OCH2CCH); 5.62 (s, 4H, –CH2–); 7.04 (s, 2H, Im–H); 7.18 (s, 2H, Im–H); 7.21 (s, 1H, Ar-H); 7.97 (s, 2H, Ar-H). 13C (100 MHz, CDCl3): 11.6 (Ar-CH3); 14.3 (–CH3); 32.1–22.8 (–(CH2)13); 49.5 (–CH2–(CH2)13–); 51.5 (–Ar-CH2N–); 56.7 (–CH2–CCH); 78.7 (–CH2CCH); 115.3 (Im1); 121.1 (Im2); 123.7 (Ar2); 122.9 (Ar1); 136.1 (–ArCCH2N–); 144.1 (Im–CH3); 158.3 (–ArCO–). MS (ESI) (m/z, %): 413.4, [(M − 2Br)/2]2+, 100%. Rf: 0.6 (DCM
:MeOH 95:5). mp: 154 °C.
4 . 1H NMR (400 MHz, CDCl3): 0.86 (t, J = 6.7 Hz, 6H, –CH3); 1.25–1.31 (m, 60H, –(CH2)15–); 1.88 (m, 4H, –NCH2CH2–); 2.71 (s, 1H, –CCH); 4.14 (t, J = 6.9 Hz, –NCH2–); 4.61 (s, 2H, –OCH2CCH); 5.20 (s, 4H, –CH2–); 6.61 (s, 2H, Ar-H); 7.09 (s, 1H, Ar-H); 7.19 (s, 2H, Im–H); 7.44 (s, 2H, Im–H). 13C NMR (100 MHz, DMSO-d6): 0.84 (t, J = 6.7 Hz, 6H, –CH3); 1.18–1.26 (m, 60H, –(CH2)15–); 1.77 (m, 4H, –NCH2CH2–); 3.53 (s, 1H, –CCH); 4.01 (m, 4H, –NCH2–); 4.63 (m, 2H, –OCH2CCH); 5.16 (s, 4H, –CH2–); 6.51 (s, 1H, Ar-H); 6.72 (s, 2H, Ar-H); 7.50 (s, 2H, Im–H); 7.58 (s, 2H, Im–H). 13C (400 MHz, CDCl3): 14.3 (–CH2(CH2)13CH3); 32.1–22.9 (–CH2(CH2)13CH3 + –CH2–); 51.7 (–CH2(CH2)13CH3); 54.2 (–Ar-CH2N–); 67.2 (–CH2CCH); 121.8 (Ar1); 123.4 (Ar2); 158.2 (–ArCO–); 191.6 (Im1 + Im2); 207.2 (Im–Au). MS (MALDI-TOF) (m/z, %): DHB-supported (993.8, [M − H − Br]+, 100%; 955.7, [M − H − Br − CH2CCH]+, 25%); no matrix (993.7, [M − Br]+, 75%; 955.7, [M − Br − CH2CCH]+, 100%). mp: 220 °C.
Synthesis of gold nanoparticles
Gold nanoparticles stabilized by imidazolium-based amphiphiles 2 or 3 were synthesized by modification of the biphasic Brust–Schiffrin method, as described elsewhere.20 Briefly, HAuCl4·3H2O solution (27 mg, 0.07 mmol) in water (10 mL) was mixed, in the dark, with a CH2Cl2 (10 mL) solution of the bis-imidazolium compound 2 or 3 (50 mg, 0.05 mmol) in an extraction funnel. The organic phase was collected in a round bottom flask, and NaBH4 (53 mg, 1.4 mmol) dissolved in water (5.5 mL) was added with vigorous stirring. The mixture was stirred at room temperature and in the dark for 4 h. The organic phase, which presented a dark red colour, was separated using an extraction funnel and the solvent was evaporated in a rotary evaporator. The residue was resuspended and washed by centrifugation with ethanol twice and acetone twice to give 2·AuNP and 3·AuNP as red solid. To store the AuNPs, the solid was dissolved in CH2Cl2 and the solutions were kept in the dark.Cell culture and antiproliferative effects
HT-29 colon carcinoma and MDA-MB-231 breast adenocarcinoma cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) high glucose (PAA) supplemented with gentamycin (50 mg L−1) and 10% (v/v) fetal calf serum (FCS) at 37 °C under a 5% CO2 atmosphere and passaged every 7 days. The antiproliferative effects were determined by the crystal violet assay as described elsewhere.15 In short: the compounds were freshly dissolved as stock solutions in dimethylformamide (DMF) and diluted with cell culture medium to concentrations in the range of x − y (0.1% v/v DMF). HT-29 and MDA-MB-231 cells were exposed to the drug containing media for a period of 72 h in an incubator (5% CO2/37 °C) and afterwards the cell biomass was determined by crystal violet staining. The IC50 values were calculated as the concentration required to reduce cell growth by 50% compared to an untreated control.Acknowledgements
This work was supported by the EU ERDF (FEDER) funds and the Spanish Government grants TEC2011-29140-C03-01/02 and TEC2014-51940-C2-1/2. The support by COST action CM1105 is acknowledged. The authors thank Dr Arancha González (ICMAB-CSIC) for her help with XRD. L. Russo thanks the Leonardo Da Vinci – Unipharma Graduates Program for a mobility grant.Notes and references
- S. Dasari and P. Bernard Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- T. M. Simon, D. H. Kunishima, G. J. Vibert and A. Lorber, Cancer Res., 1981, 41, 94–97 CAS.
- C. K. Mirabelli, R. K. Johnson, C. M. Sung, L. Faucette, K. Muirhead and S. T. Crooke, Cancer Res., 1985, 45, 32–39 CAS.
- J. C. Lima and L. Rodriguez, Adv. Anticancer Agents Med. Chem., 2011, 11, 921–928 CrossRef CAS.
- E. Vergara, E. Cerrada, A. Casini, O. Zava, M. Laguna and P. J. Dyson, Organometallics, 2010, 29, 2596–2603 CrossRef CAS.
- C. H. Chui, R. S. M. Wong, R. Gambari, G. Y. M. Cheng, M. C. W. Yuen, K. W. Chan, S. W. Tong, F. Y. Lau, P. B. S. Lai, K. H. Lam, C. L. Ho, C. W. Kan, K. S. Y. Leung and W. Y. Wong, Bioorg. Med. Chem., 2009, 17, 7872–7877 CrossRef CAS PubMed.
- J. E. Biaglow and R. A. Miller, Cancer Biol. Ther., 2005, 4, 6–13 CAS.
- K. F. Tonissen and G. D. Trapani, Mol. Nutr. Food Res., 2009, 53, 87–103 CAS.
- P. Nguyen, R. T. Awwad, D. D. K. Smart, D. R. Spitz and D. Gius, Cancer Lett., 2006, 236, 164–174 CrossRef CAS PubMed.
- P. J. Barnard and S. J. Berners-Price, Coord. Chem. Rev., 2007, 251, 1889–1902 CrossRef CAS.
- M. V Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2006, 6, 3708–3715 RSC.
- J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 CrossRef CAS PubMed.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 CrossRef CAS PubMed.
- R. Rubbiani, S. Can, I. Kitanovic, H. Alborzinia, M. Stefanopoulou, M. Kokoschka, S. Mönchgesang, W. S. Sheldrick, S. Wölfl and I. Ott, J. Med. Chem., 2011, 54, 8646–8657 CrossRef CAS PubMed.
- W. Liu, K. Bensdorf, M. Proetto, U. Abram, A. Hagenbach and R. Gust, J. Med. Chem., 2011, 54, 8605–8615 CrossRef CAS PubMed.
- J. Arcau, V. Andermark, M. Rodrigues, I. Giannicchi, L. Pérez-García, I. Ott and L. Rodríguez, Eur. J. Inorg. Chem., 2014, 2014, 6117–6125 CrossRef CAS.
- A. Pratesi, C. Gabbiani, E. Michelucci, M. Ginanneschi, A. M. Papini, R. Rubbiani, I. Ott and L. Messori, J. Inorg. Biochem., 2014, 136, 161–169 CrossRef CAS PubMed.
- A. Citta, E. Schuh, F. Mohr, A. Folda, M. L. Massimino, A. Bindoli, A. Casini and M. P. Rigobello, Metallomics, 2013, 5, 1006–1015 RSC.
- L. Casal-Dujat, M. Rodrigues, A. Yagüe, A. C. Calpena, D. B. Amabilino, J. González-Linares, M. Borrás and L. Pérez-García, Langmuir, 2012, 28, 2368–2381 CrossRef CAS PubMed.
- L. Casal-Dujat, P. C. Griffiths, C. Rodríguez-Abreu, C. Solans, S. Rogers and L. Pérez-García, J. Mater. Chem. B, 2013, 1, 4963–4971 RSC.
- E. Amirthalingam, M. Rodrigues, L. Casal-Dujat, A. C. Calpena, D. B. Amabilino, D. Ramos-López and L. Pérez-García, J. Colloid Interface Sci., 2015, 437, 132–139 CrossRef CAS PubMed.
- M. Rodrigues, A. C. Calpena, D. B. Amabilino, D. Ramos-López, J. de Lapuente and L. Pérez-García, RSC Adv., 2014, 4, 9279–9287 RSC.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- C. He, L. W. Li, W. D. He, W. X. Jiang and C. Wu, Macromolecules, 2011, 44, 6233–6236 CrossRef CAS.
- E. Schuh, S. M. Valiahdi, M. A. Jakupec, B. K. Keppler, P. Chiba and F. Mohr, Dalton Trans., 2009, 48, 10651–10659 Search PubMed.
- J. Arcau, V. Andermark, E. Aguiló, A. Gandioso, A. Moro, M. Cetina, J. C. Lima, K. Rissanen, I. Ott and L. Rodríguez, Dalton Trans., 2014, 43, 4426–4436 RSC.
- M. Matmor and N. Ashkenasy, J. Mater. Chem., 2011, 21, 968–974 RSC.
- Z. Huo, C.-K. Tsung, W. Huang, X. Zhang and P. Yang, Nano Lett., 2008, 8, 2041–2044 CrossRef CAS PubMed.
- M. P. Casaletto, A. Longo, A. Martorana, A. Prestianni and A. M. Venezia, Surf. Interface Anal., 2006, 38, 215–218 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21621d |
| ‡ Present address: School of Pharmacy, The University of Nottingham, University Park, Nottingham NG72RD, England, UK. |
| This journal is © The Royal Society of Chemistry 2016 |