
Triterpenes from the fruits of Rosa laevigata with acetylcholinesterase and Aβ-aggregation inhibitory activities†
Pin-Yi Gaoab,
Meng Wanga,
Xue-Gui Liu*a,
Yi-Xing Gaoa,
Jia-Luo Lia,
Zhen-Xue Zhanga,
Hou-Wen Linc and
Shao-Jiang Song*b
aCollege of Pharmaceutical and Biotechnology Engineering, Shenyang University of Chemical Technology, Shenyang 110142, P. R. China. E-mail: liuxuegui72@163.com; gpy1981@163.com
bKey Laboratory of Structure-Based Drug Design and Discovery, Ministry of Education, School of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, Shenyang 110016, P. R. China. E-mail: songsj99@163.com
cKey Laboratory for Marine Drugs, Department of Pharmacy, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200127, P. R. China
First published on 23rd December 2015
Abstract
A lupane-type triterpene (1) featuring a rare 2-hemiacetal moiety in its A ring and aromatic ester derivatives (2–6) were isolated from the fruits of R. laevigata and evaluated to possess acetylcholinesterase and Aβ-aggregation inhibitory activities. Their structures were determined by multi-spectroscopic methods, chemosynthesis and CD analysis.
Acetylcholinesterase (AChE) plays a key role in the treatment of Alzheimer's disease (AD)1,2 and it has been suggested that a decreased concentration of ACh appears to be a critical element in the development of dementia of AD.3 Due to the correlation of AD with a cholinergic deficit, AChE inhibitors from herbal drugs or herbal medicinal preparations have gained a lot of scientific interest.4 In addition, AChE is known to induce amyloid-β (Aβ) formation leading to the highly toxic AChE–Aβ peptide complexes.5 And recent studies have shown that some small molecule therapies exhibit AChE inhibition as well as reduce the formation of neurotoxic Aβ-aggregates.6
In our screening study on new AChE inhibitors from plant resources, we have indicated that Rosa laevigata Michx exerted AChE inhibitory activity, thus keeping the normal transmission of nerve impulses. Rosa laevigata Michx, an evergreen climbing shrub, is widely distributed in China. As a famous medicinal plant, the roots, fruits, flowers and leaves of R. laevigata have been recorded as traditional medicines.7–9 In this communication, we describe the isolation, structure determination of compounds 1–6 (Fig. 1), as well as AChE and Aβ-aggregation inhibitory activities by in vitro assays and molecular modeling studies.
 | ||
| Fig. 1 Structure of compounds 1–6. | ||
Exhaustive extraction of the dried fruits of R. laevigata (10 kg) with 70% ethanol (60 L) followed by reflux three times (80–85 °C), concentration in vacuo and then fractionation by dichloromethane five times (CH2Cl2 extract 128.3 g, 1.28%). This was subjected to column chromatography using CH2Cl2/MeOH (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → 5:1) solvent to gain fractions E1–E7 and the second fraction (E2) was purified by reversed-phase C18 silica gel using MeOH/H2O (60:40 → 90:10) to give compounds 1 (12 mg) and 5 (36 mg). E3 (9.8 g) and E4 (11.2 g) were applied to a column of MCI gel MeOH/H2O (30:70 → 100:0) respectively. The major fractions of Frs. E3 finally performed by reversed-phase C18 silica gel using MeOH/H2O with gradient solvent system of (50:50 → 90:10) and HPLC to isolate compounds 2 (16 mg), 6 (13 mg). Compounds 3 (11 mg), 4 (38 mg) were obtained from Frs. E4 by the similar way.
:1 → 5:1) solvent to gain fractions E1–E7 and the second fraction (E2) was purified by reversed-phase C18 silica gel using MeOH/H2O (60:40 → 90:10) to give compounds 1 (12 mg) and 5 (36 mg). E3 (9.8 g) and E4 (11.2 g) were applied to a column of MCI gel MeOH/H2O (30:70 → 100:0) respectively. The major fractions of Frs. E3 finally performed by reversed-phase C18 silica gel using MeOH/H2O with gradient solvent system of (50:50 → 90:10) and HPLC to isolate compounds 2 (16 mg), 6 (13 mg). Compounds 3 (11 mg), 4 (38 mg) were obtained from Frs. E4 by the similar way.
Compound 1 was obtained as colorless needles. The molecular formula was established as C30H48O4 by HRESIMS, which showed a sodium adduct ion peak [M + Na]+ at m/z 495.3478 (calculated 495.3445). The 1H NMR spectrum (Table 1) of 1 showed signals due to six tertiary methyl groups at δH 0.85, 0.97, 1.02, 1.06 × 2 and 1.72, two olefinic protons at δH 4.54 (1H, br s), 4.63 (1H, br s), two oxygen-bearing methylene protons at δH 3.04 (1H, d, J = 13.0 Hz), 3.09 (1H, d, J = 13.0 Hz), as well as one hemiacetal proton at δH 5.01 (1H, dd, J = 10.0, 5.0 Hz). The 13C NMR spectrum exhibited one carboxylic acid group at δC 181.0, one 1,1-disubstituted double bond at δC 110.3 and 151.9, one oxygen-bearing methylene carbon at δC 71.7, and one hemiacetal carbon at δC 94.9. On account of the molecular formula C30H48O4, the degree of unsaturation of 1 was seven including one carbonyl and one olefinic functionality. Thus, the number of rings of 1 should be five. Further information about the planar skeleton structure of 1 was obtained from the HSQC and HMBC experiments (Fig. 2). The HMBC spectrum exhibited correlations from δH 0.85 (23-Me) to C-3, C-4, C-5, C-24; δH 0.97 (24-Me) to C-3, C-4, C-5, C-23; δH 1.06 (25-Me) to C-1, C-5, C-9, C-10; δH 1.06 (26-Me) to C-7, C-8, C-9, C-14; δH 1.02 (27-Me) to C-13, C-14, C-15; δH 1.72 (30-Me) to C-19, C-20, C-29; δH 5.01 (H-2) to C-3; δH 3.04, 3.09 (H2-3) to C-2, C-4, C-5, C-23, C-24; δH 0.89 (H-5) to C-4, C-6, C-7, C-10, C-23, C-24; δH 1.31 (H-9) to C-1, C-8, C-10, C-11, C-26; δH 2.39 (H-13) to C-12, C-15, C-17, C-18, C-27; δH 3.07 (H-19) to C-18, C-20, C-21, C-29, C-30, indicating the planar structure of the triterpene skeleton. All above spectroscopic data suggested that 1 was closely comparable to a lupane-type triterpene except the A ring featuring a rare 2-hemiacetal moiety.
| Position | Compound 1 | Compound 2 | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1 | 1.36 dd (15.0, 10.0) | 46.7 | 0.99 dd (12.5, 10.0) | 44.6 |
| 2.13 dd (15.0, 5.0) | 2.01 dd (12.5, 4.5) | |||
| 2 | 5.01 dd (10.0, 5.0) | 94.9 | 5.03 td (10.0, 4.5) | 74.3 |
| 3 | 3.04 d (13.0) | 71.7 | 3.21 d (10.0) | 79.2 |
| 3.09 d (13.0) | ||||
| 4 | 39.9 | 40.7 | ||
| 5 | 0.89 t like (6.0) | 61.7 | 0.87 m | 55.5 |
| 6 | 1.51 m | 21.8 | 1.33 m | 18.9 |
| 1.50 m | ||||
| 7 | 1.46 m | 35.6 | 1.37 m | 34.7 |
| 8 | 42.6 | 41.3 | ||
| 9 | 1.31 m | 48.8 | 1.35 m | 50.9 |
| 10 | 42.0 | 39.0 | ||
| 11 | 1.34 m | 23.6 | 1.15 m | 21.5 |
| 1.59 m | 1.31 m | |||
| 12 | 1.09 m | 27.4 | 0.97 m | 25.9 |
| 1.78 m | 1.67 m | |||
| 13 | 2.39 td (12.0, 3.5) | 39.9 | 2.23 td (12.0, 3.0) | 38.5 |
| 14 | 43.9 | 43.0 | ||
| 15 | 1.33 m | 30.6 | 1.09 m | 30.1 |
| 1.56 m | 1.42 m | |||
| 16 | 2.27 m | 33.4 | 1.37 m | 32.6 |
| 2.12 m | ||||
| 17 | 57.6 | 56.4 | ||
| 18 | 1.67 t like (11.5) | 50.5 | 1.51 t like (11.0) | 49.5 |
| 19 | 3.07 m | 48.6 | 2.93 m | 47.5 |
| 20 | 151.9 | 151.2 | ||
| 21 | 1.44 m | 31.7 | 1.11 m | 31.1 |
| 1.99 m | 1.34 m | |||
| 22 | 1.49 m | 38.1 | 1.41 m | 37.3 |
| 1.95 m | 1.81 m | |||
| 23 | 0.85 s | 28.4 | 0.98 s | 29.3 |
| 24 | 0.97 s | 21.4 | 0.78 s | 17.7 |
| 25 | 1.06 s | 15.0 | 0.93 s | 17.8 |
| 26 | 1.06 s | 17.6 | 0.88 s | 16.6 |
| 27 | 1.02 s | 14.9 | 0.94 s | 15.3 |
| 28 | 181.0 | 180.3 | ||
| 29 | 4.54 br s | 110.3 | 4.53 br s | 110.4 |
| 4.63 br s | 4.66 br s | |||
| 30 | 1.72 s | 19.5 | 1.62 s | 19.8 |
| 1′ | 166.5 | |||
| 2′ | 131.5 | |||
| 3′ | 7.97 dd (8.0, 1.0) | 130.1 | ||
| 4′ | 7.50 t (8.0) | 120.3 | ||
| 5′ | 7.62 t (8.0) | 133.8 | ||
| 6′ | 7.50 t (8.0) | 129.3 | ||
| 7′ | 7.97 dd (8.0, 1.0) | 130.1 | ||
 | ||
| Fig. 2 Selected HMBC correlations of 1 (left). Selected NOESY correlations and relative configuration of 1 (right). | ||
The relative configuration of 1 was confirmed by the 1H NMR coupling constants and the correlations in its NOESY plot (Fig. 2). The vicinal coupling constant J(1,2) of 10.0 Hz suggested a cis relationship between the protons Ha-1 and H-2. To the crucial NOE correlations between H-5/H-9, H-9/H3-27, H-18/H3-27 and H-18/H3-30 suggested that they were oriented on the same side of ring A–E. Furthermore, H3-25 showed NOE correlations with H-2 and H3-26, H-13 showed NOE correlation with H3-26 and H-19, suggesting that these protons were on the same face of ring A–E. The absence of cross-peaks between δH 0.89 (H-5) and δH 5.01 (H-2) indicated that these two groups of H-2/H-13/H-19/H3-25/H3-26 and H-5/H-9/H-18/H3-27/H3-30 were oriented on the opposite side.
The absolute configuration of 1 was established by the CD exciton chirality method and comparison of the experimental ECD spectrum and calculated ECD data.10,11 In order to use the signs of the Cotton effects as indicators according to published protocols,12 the 2-O-benzoyl derivative (1a) (Fig. 3) of 1 was synthesized following the method described in the ESI (S.1.8.†). The CD spectrum of 1a (Fig. 4) showed a positive Cotton effect, which centered around the UV maximum at 220 nm, corresponding to the exciton coupling between the chromophores of the benzoate moiety at C-2 (225 nm, Δε + 14.05) and the Δ20,29 double bond (211 nm, Δε − 11.60). The counter-clockwise spatial geometry of the two chromophores, thus showed that the absolute configuration of 1 was 2S, 5S, 8R, 9S, 10S, 13R, 14R, 17S, 18R and 19R (Fig. 2), which was in agreement with that defined by comparison of the experimental ECD spectrum and calculated ECD data using the time-dependent density functional theory (TD-DFT) method at the B3LYP/6-31G (d, p) level (Fig. 4).
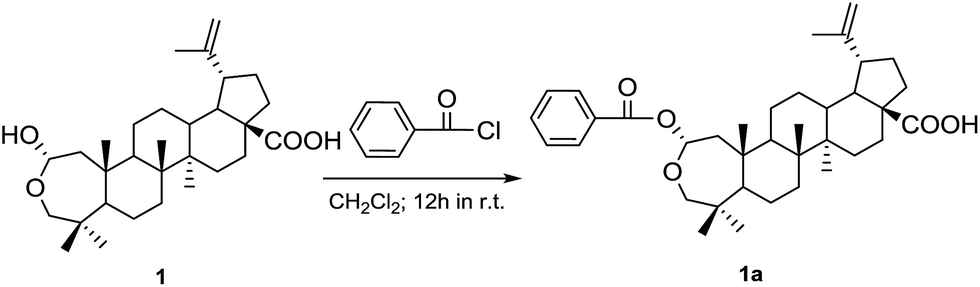 | ||
| Fig. 3 Synthesis of benzoyl ester 1a of Laevigaterpene A (1). | ||
 | ||
| Fig. 4 Experimental ECD (solid line) and calculated ECD (dotted line) spectra for 1 and 2. | ||
Compound 2 was obtained as colorless needles. The molecular formula was established as C37H52O5 by HRESIMS, which showed a sodium adduct ion peak [M + Na]+ at m/z 599.3719 (calculated 599.3707). The 1H NMR spectrum (Table 1) of 2 showed signals due to six tertiary methyl groups at δH 0.78, 0.88, 0.93, 0.94, 0.98, and 1.62, two olefinic protons at δH 4.53 (1H, br s), 4.66 (1H, br s), two oxygen-bearing methylene protons at δH 3.21 (1H, d, J = 10.0 Hz), 5.03 (1H, dd, J = 10.0, 4.5 Hz), and five aromatic protons of a single phenyl ring (single substitution pattern) at δH 7.97 (2H, dd, J = 8.0, 1.0 Hz), 7.50 (2H, t, J = 8.0 Hz), 7.62 (1H, t, J = 8.0 Hz). The 13C NMR spectrum exhibited two carboxyl carbons at δC 180.3, 166.5, one 1,1-disubstituted double bond at δC 110.4 and 151.2, two oxygen-bearing methylene carbons at δC 74.3, 79.2 and five aromatic carbons of a single phenyl ring at δC 129.3 × 2, 130.1 × 2, 131.5 and 133.8. Further information about the planar skeleton structure of 2 was obtained from the HSQC and HMBC experiments (ESI, Fig. 12, 13†). The correlations in HMBC spectrum from δH 0.98 (23-Me) to C-3, C-4, C-5, C-24; δH 0.78 (24-Me) to C-3, C-4, C-5, C-23; δH 0.93 (25-Me) to C-1, C-5, C-9, C-10; δH 0.88 (26-Me) to C-7, C-8, C-9, C-14; δH 0.94 (27-Me) to C-13, C-14, C-15; δH 1.62 (30-Me) to C-19, C-20, C-29; δH 0.87 (H-5) to C-4, C-6, C-7, C-10, C-23, C-24; δH 1.35 (H-9) to C-1, C-8, C-10, C-11, C-26; δH 2.23 (H-13) to C-12, C-15, C-17, C-18, C-27; δH 2.93 (H-19) to C-18, C-20, C-21, C-29, C-30, suggested that 2 was also possessing a lupane-type skeleton. Moreover, the key correlation from δH 5.03 (H-2) to one ester carbonyl at δC166.5 in HMBC suggested that a benzoyloxy was likely located at C-2. The CD spectrum of 2 (Fig. 4) showed a positive Cotton effect, which centered around the UV maximum at 225 nm, corresponding to the exciton coupling between the chromophores of the benzoate moiety at C-2 (229 nm, Δε + 4.58) and the Δ20,29 double bond (211 nm, Δε + 2.17). The counter-clockwise spatial geometry of the two chromophores, thus showed that the absolute configuration of 2 was 2R, 5S, 8R, 9S, 10S, 13R, 14R, 17S, 18R and 19R. In addition, this assignment is in good conformity with the result of a combined ECD strategy of experiment and calculation. The experimental ECD spectrum of 2 matched well with the calculated ECD data (Fig. 4).
In the course of searching for AChE inhibitors from herb medicines, the total EtOH extract and CH2Cl2-soluble extract fraction from the fruits of R. laevigata were found to exhibit AChE inhibitory activity, with IC50 values of 36.3 μg mL−1 and 26.4 μg mL−1 (Fig. 5). Further multi-column chromatographic fractionation led to obtain six pentacyclic triterpenes, including lupinane (1–4) and oleanane type (5, 6). Their IC50 values for AChE inhibition are shown in Table S3.† Among them, compounds 1 (IC50 14.4 μg mL−1) and 4 (IC50 15.2 μg mL−1) showed stronger activity than others (Fig. 5). Since compounds 3 and 4 are stereoisomers, the observed differences in the inhibition of AChE could only be due to different spatial configurations. Compound 1, with uncommon hemiacetal skeleton at A ring compared to 2–4, showed strongest activity of all the six isolates. These relationships of structures and activities suggested that the configuration and A ring have important effect on AChE inhibition. In addition, the different structure at E ring between lupinane (1–4) and oleanane type (5, 6) could be another factor.
 | ||
| Fig. 5 AChE inhibition activity of compounds 1–6, positive control and different fractions. | ||
In parallel with the AChE inhibition assay, the new active inhibitors were screened for their inhibitory activity on Aβ self-aggregation by the thioflavin T competition method.13 The anti-aggregating activity of six isolates along with reference compound trolox are presented in Table S4.† At a concentration of 100 μM, compounds 1–6 and trolox exhibited varying degrees of inhibition (inactive to 65.2 ± 10% inhibition of aggregation).
Among the six compounds, the enzyme-ligand binding interactions of the more potent AChE inhibitors 1 and 4 were investigated by molecular modeling studies14,15 (Fig. 6). These investigations indicate that (a) The 2,3-OH of 1 were in close proximity to the catalytic residues (Ser122 and Tyr121) and were undergoing hydrogen bonding interactions (distance 2.59, 2.77 and 3.24 Å). Three hydrogen bonding interactions were also oriented between the Tyr121 Glu199 and 2, 3-OH and 30-COOH of 4 (distance 2.31, 2.78 and 3.39 Å). (b) Both 1 and 4 were oriented in an aromatic pocket comprised of Tyr334, Phe331, Phe330 and Trp84. In addition, Gly441, Gly117, Tyr130, and Tyr70 are also considered as key active residues interacting with the natural ligands.
 | ||
| Fig. 6 Docking of 1 (A and B) and 4 (C and D) in the active site of AChE. Green dotted lines represent hydrogen bonding. Hydrogen atoms are not shown for clarity. | ||
The cytotoxicity of compounds 1–6 was evaluated using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) – based colorimetric assay. The cell viability of SH-SY5Y neuroblastoma cells after exposure to 1–6 was compared with reference compound galanthamine (Table S5†). At 40 μM,6 the compounds 2, 5, 6 that exhibited AChE inhibition (IC50 = 27.5, 28.5, 25.2 μg mL−1) were nontoxic (98%, 96%, 97% cell viability). For comparison, the viability of SH-SY5Y cells was 96% in the presence of the reference compound galanthamine under the same experimental conditions. In addition, the compounds 1, 3 and 4 were only slightly toxic under these conditions, with cell viabilities of 75%, 80% and 85%, respectively. These studies indicate that compounds 1–6 were nontoxic or only slightly toxic to SH-SY5Y cells.
Conclusions
In summary, the work discovered a novel skeleton compound laevigaterpene A (1) and a new compound laevigaterpene B (2), together with four known ones (3–6) from the fruits of R. laevigata. Extensive spectroscopic analyses were employed for structure elucidation, especially, the application of the CD exciton chirality coupled with the calculated ECD data have provided a better understanding of the absolute configuration of lupinane type triterpenes. Moreover, we found that these isolates exhibit both AChE and Aβ-aggregation inhibitory activities which are agree with docking studies. In aggregation, a novel small molecule (1) and aromatic ester derivatives (2–6) has been characterized and verified to two functions in the condition of nontoxic or only slightly toxic which might have a possibility for use in the multitarget for AD therapy.Acknowledgements
Financial support from the Project of Innovation Team (LT2015027) of Liaoning of P. R. China, Foundation (L2015419) from the Project of Education Department of Liaoning province of P.R. China, Financial support of the National Funds for Distinguished Young Scientists (81225023), STCM project (13401900802), the National Science & Technology Major Project (2013ZX09103002-017), and Financial supported by Program for National Undergraduate Training Programs for Innovation and Entrepreneurship (2015101490979) are gratefully acknowledged. The authors would like to thank Molegro ApS for kindly providing us with a free evaluation of their software packages and associate prof. Jian Wang of Shenyang Pharmaceutical University for docking studies.Notes and references
- E. Oueis, F. Nachon, C. Sabot and P. Y. Renard, Chem. Commun., 2014, 50, 2043–2045 RSC.
- E. Oueis, G. Santoni, C. Ronco, O. Syzgantseva, V. Tognetti, L. Joubert, A. Romieu, M. Weik, L. Jean, C. Sabot, F. Nachone and P. Y. Renard, Org. Biomol. Chem., 2014, 12, 156–161 CAS.
- Y. Chen, X. L. Xu, T. M. Fu, W. Li, Z. L. Liu and H. P. Sun, RSC Adv., 2015, 5, 90288–90294 RSC.
- H. R. Adhami, V. Fitz, A. Lubich, H. Kaehlig, M. Zehl and L. Krenn, Phytochem. Lett., 2014, 10, lxxxii–lxxxvii CrossRef CAS.
- F. Belluti, A. Rampa, L. Piazzi, A. Bisi, S. Gobbi, M. Bartolini, V. Andrisano, A. Cavalli, M. Recanatini and P. J. Valenti, J. Med. Chem., 2005, 48, 4444–4456 CrossRef CAS PubMed.
- T. Mohamed, X. B. Zhao, L. K. Habib, J. Yang and P. P. N. Rao, Bioorg. Med. Chem., 2011, 9, 2269–2281 CrossRef PubMed.
- N. Zeng, Y. Shen, L. Z. Li, W. H. Jiao, P. Y. Gao, S. J. Song, W. S. Chen and H. W. Lin, J. Nat. Prod., 2011, 74, 732–738 CrossRef CAS PubMed.
- G. Q. Yan, S. P. Li, J. Hu, X. Y. Zhai, W. Ma, N. Li and K. J. Wang, Biochem. Syst. Ecol., 2014, 52, 23–26 CrossRef CAS.
- S. J. Choi, M. J. Kim, H. J. Heo, H. K. Kim, B. Hong, C. J. Kim, B. G. Kim and D. H. Shin, Amyloid, 2006, 13, 6–12 CrossRef PubMed.
- N. Harada, J. Iwabuchi, Y. Yokota, H. Uda and K. J. Nakanishi, J. Am. Chem. Soc., 1981, 103, 5590–5591 CrossRef CAS.
- N. Berova, K. Nakanishi and R. W. Woody, in Circular Dichroism: Principles and Applications, Wiley-VCH, New York, 2000, pp. 337–382 Search PubMed.
- H. D. Chen, S. P. Yang, X. F. He, H. B. Liu, J. Ding and J. M. Yue, Tetrahedron, 2010, 66, 5065–5070 CrossRef CAS.
- K. H. Chang, Y. J. Chiu, S. L. Chen, C. H. Huang, C. H. Lin, T. H. Lin, C. M. Lee, C. H. Wu, C. C. Huang, H. C. Fung, Y. C. Chen, J. Y. Lin, C. F. Yao, H. J. Huang, G. J. Lee-Chen, M. C. Lee and H. M. Hsieh-Li, Neuropharmacology, 2016, 101, 309–319 CrossRef CAS PubMed.
- C. S. Azad, V. M. Balaramnavar, I. A. Khan, P. K. Doharey, J. K. Saxena and A. K. Saxena, RSC Adv., 2015, 5, 82208–82214 RSC.
- H. M. Greenblatt, C. Guillou, D. Guenard, A. Argaman, S. Botti, B. Badet, C. Thal, I. Silman and J. L. Sussman, J. Am. Chem. Soc., 2004, 126, 15405–15411 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR, HRESIMS, ECD spectra and docking studies and detailed experimental procedures. See DOI: 10.1039/c5ra21590k |
| This journal is © The Royal Society of Chemistry 2016 |