
Scalable synthesis of nanometric α-Fe2O3 within interconnected carbon shells from pyrolytic alginate chelates for lithium storage†
Abstract
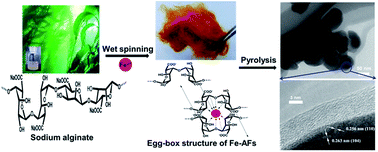
Nanostructured α-Fe2O3 with a carbon coating has advantages over commercially available graphitic anodes in lithium ion batteries, including high theoretical anodic capacity (1007 mA h g−1), low cost and environmental safety. However, parts of its merits were cancelled out by the current route used for its synthesis. For example, most of its synthesis pathways are tedious, costly, on small scale and environmentally unfriendly. By combining two naturally abundant materials (iron ions and sodium alginate) into conventional wet-spinning and pyrolysis processes, we show that α-Fe2O3 could be nanostructured and carbon-coated using a facile and yet scalable technique. The successful synthesis of nanostructured α-Fe2O3 within interconnected carbon shells strongly depends on the chelation of iron ions and alginate into “egg-box” networks and an optimized pyrolysis step. Due to an extraordinary combination of nanometric scale, improved electrical conductivity and spatial confinement of the carbon coating, the resultant materials exhibit a high lithium storage capacity (e.g. up to ∼1400 mA h g−1 at a current of 50 mA g−1 in the initial cycle) and great stability (e.g. ∼560 mA h g−1 after 600 cycles at 200 mA g−1) as well as a coulombic efficiency of 97.5%. Owing to the cost-efficient, environmentally friendly and scalable production, this synthesis may pave a highly promising way to the macroscopic preparation of α-Fe2O3/carbon hybrid materials as anodes in lithium ion batteries.
 Please wait while we load your content...
Please wait while we load your content...

