
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Precision microfilters as an all in one system for multiplex analysis of circulating tumor cells†
Daniel L.
Adams
*a,
R. Katherine
Alpaugh
b,
Stuart S.
Martin
c,
Monica
Charpentier
c,
Saranya
Chumsri
cd,
Massimo
Cristofanilli
e,
Diane K.
Adams
f,
Olga V.
Makarova
g,
Peixuan
Zhu
h,
Shuhong
Li
h,
Cha-Mei
Tang
h and
Steingrimur
Stefansson
i
aCreatv MicroTech, Inc., 1 Deer Park Dr., Monmouth Junction, NJ 08852, USA. E-mail: dan@creatvmicrotech.com
bFox Chase Cancer Center, Protocol Support Laboratory, 333 Cottman Ave., Philadelphia, PA 19111, USA
cUniversity of Maryland Baltimore Greenebaum Cancer Center, 655 W. Baltimore St., Baltimore, MD 21136, USA
dMayo Clinic Cancer Center, 4500 San Pablo Rd., Jacksonville, FL 32224, USA
eRobert H Lurie Comprehensive Cancer Center, Northwestern University, 645 N Michigan Avenue, Chicago, IL 60611, USA
fRutgers, the State University of New Jersey, 71 Dudley Rd., New Brunswick, NJ 08901, USA
gCreatv MicroTech, Inc., 2242 West Harrison St., Chicago, IL 60612, USA
hCreatv MicroTech, Inc., 9900 Belward Campus Dr. Rockville, MD 20850, USA
iHememics Biotechnologies, 12111 Parklawn Dr. Rockville, MD 20852, USA
First published on 11th January 2016
Abstract
Enumeration of circulating tumor cells (CTCs) from cancer patient blood is an established diagnostic assay used to evaluate patient status as a singleplex test. However, in the coming age of personalized medicine, multiplex analysis of patient CTCs, including proteomic and genomic techniques, will have to be integrated with CTC isolation platform technologies. Advancements in microfabrication have demonstrated that CTCs can be isolated and analyzed using microfluidic lab-on-a-chip devices. However, to date, most microfluidic devices are either still in the development phase, not applicable to all clinical tests, or are not commercially available. To overcome these discrepancies, we describe an all-in-one device for the isolation and multiplexing of clinically applicable CTC assays. Microfilters present an ideal lab-on-a-chip platform for analysis of CTCs as non-toxic and inert materials allow for a multitude of tests from cell growth through clinical staining techniques, all without background interference. Lithographically fabricated microfilters, can be made with high porosity, precise pore dimensions, arrayed pore distribution, and optimized for CTC size-based isolation. In this study we describe microfilter use in isolation and in situ analysis of CTCs using multiple sequential techniques including culture, FISH, histopathological analysis, H&E staining, photobleaching and re-staining. Further, as a proof of principle, we then describe the ability to quantitatively release patient derived CTCS from the microfilters for potential use in downstream genomic/proteomic analysis.
Introduction
The primary mechanism of metastasis is believed to be the extravasation, or shedding, of cancerous epithelial cells into circulation. These circulating tumor cells (CTCs) can travel throughout the body, adhere to vascular beds of organs, infiltrate, grow and impair organ function. However, according to animal studies, this process is very inefficient, as <1% of shed CTCs are viable after 24 hours intravasation into circulation, despite ∼107 tumor cells being shed into the bloodstream per gram of tumor tissue every 24 hours.1–3 This lack of viability could be due to apoptosis through anoikis, actions of natural killer cells of the host, or shear stress from capillary flow.4,5 In accordance with animal studies, few CTCs (∼1 CTC per 7.5 ml blood sample) are found in early stage cancer patients, but as the cancer progresses, the CTC count can increase to >5 CTCs per 7.5 ml blood sample.5–9 Therefore, technologies that can capture and analyze these rare cells from patient blood samples are being vigorously pursued for diagnostic purposes, and to determine whether prescribed therapies are effective.Strategies for isolating CTCs from whole blood samples generally fall into two broad categories (1) affinity based isolation and (2) label-free isolation. The only clinically accepted method for enumerating CTCs from cancer patients is the CellSearch® CTC test, which is an affinity based isolation of CTCs using magnetic nanoparticles coated with antibodies against the epithelial cell adhesion molecule (EpCAM).6,8,10,11 The captured cells are then further characterized by the staining of anti-cytokeratin (CK) antibodies and the non-staining with anti-CD45 (leukocyte common antigen) antibodies. Although the CellSearch® CTC test is clinically validated, it is not designed for CTC analysis beyond enumeration of basic biomarker expression and therefore has limited personalized medicine applications.5,9,12
Alternatives to CellSearch® that are more adaptable to downstream analysis of CTCs, post-isolation are actively being pursued.13–16 These label-free CTC isolation techniques (i.e. size based isolation, buffy coat smears, microfluidics, electrophoretic, etc.) are not limited by markers expressed on the circulating cells and have been reported to isolate far greater numbers of CK+ expressing cells from cancer patient blood samples than the CellSearch® system, at times numbering thousands of CK+, or EpCAM+ cells per milliliter of patient blood.6,12,17–19 Although the greater number of CK+ cells isolated by these techniques can be attributed to greater isolation efficiency, the clinically validated data provided by CellSearch® has only been recently reproduced by filtration methodology.6,12,20 Additionally, like CellSearch®, many of these CTC isolation systems also have limiting downstream applications, i.e. blood smears cannot be cultured; microfluidics have limited blood volume inputs and long run times; and electrophoretic techniques requires additional purification.12–17,20–22
It has been postulated that more detailed examinations of CTCs will yield clinically important data. However, currently, there is no single commercially available lab on a chip platform that can isolate and sequentially analyze CTCs in situ using multiple methods. To date, the only label-free CTC assay that shows both increased sensitivity as well as correlation to the clinically validated CellSearch® test is the CellSieve™ system.5,6,12,20 Since CellSieve™ filters can identify the clinically relevant CTCs, without using magnetic particles that can obscure cellular details; we have been actively assessing techniques that can further characterize CTCs immobilized on the microfilters. The ability to expand CTC analysis beyond single plex enumeration would greatly extend utility of CTCs to include more than basic prognostication. The ability to expand CTC analysis beyond single plex enumeration would greatly extend utility of CTCs to include more than basic prognostication.
CellSieve™ filters are not autofluorescent and are biologically inert, allowing for staining of captured cells using multiple fluorescent antibodies and for the potential growth of growing captured cells on the filters.18,23,24 Here we suggest that cells captured on this platform can be cultured in situ or harvested using a backwash procedure, and subsequently analyzed using numerous downstream applications (Fig. 1). We show that the platform allows for multiple downstream techniques, such as culture, histopathological subtyping, fluorescent in situ hybridization (FISH), scanning electron microscopy (SEM) and single cell capture using micromanipulation. Our data supports the view that CTC capture, culture and clinically applicable testing are possible using a size based all-in-one lab-on-chip platform capable of analyzing and characterizing CTC biology.
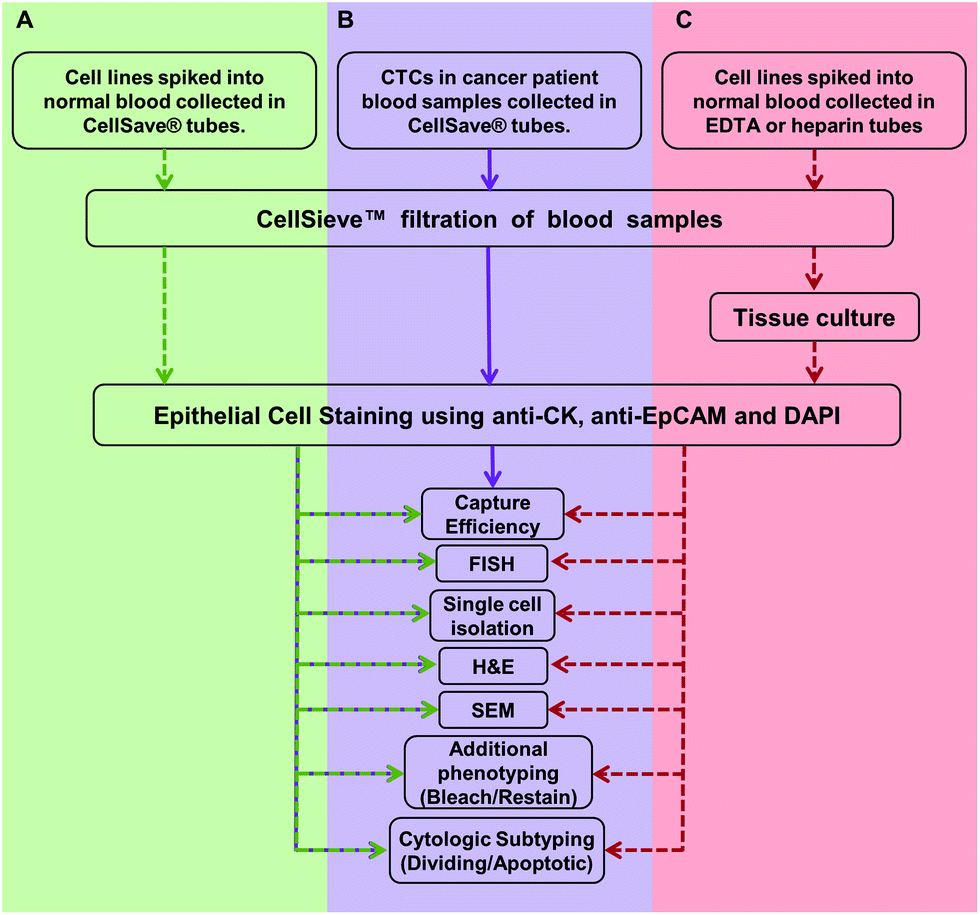 | ||
| Fig. 1 Overview of the work flow and methodologies described in this study. (A) Assays are developed and optimized with cancer cell lines spiked into normal blood samples (green). Cancer cell lines are spiked into blood samples collected in CellSave® tubes. The sample is filtered and CTCs are identified using presence of anti-cytokeratin and anti-EpCAM, with absence of anti-CD45. CTCs are quantified, then stained by FISH, H&E, etc. (B) Assays are run on patient samples (purple). Blood from cancer patients were collected in CellSave® tubes, filtered and CTCs were identified. CTCs were counted and the clinically useful subtypes were quantified (i.e. CTCs in division, apoptotic CTCs, etc.).6 Many of these cells were further subtyped by FISH or H&E stain (Fig. 2). (C) Proof of principal assay for expansion of CTCs. Viable cancer cell lines were spiked into normal blood collected in either EDTA and isolated by filtration. The filter bound cells were then expanded in culture media for eventual use in other models.24,25 Dotted arrow indicates that cell lines were used. Solid lines indicate the assay was developed with cell lines and has proceeded onto patient samples. | ||
Materials and methods
Healthy and patient blood samples used for these studies
Sixteen anonymized cancer patient peripheral blood samples, breast (n = 10) or prostate (n = 6), collected in CellSave tubes were supplied through a collaborative agreement with Fox Chase Cancer Center (FCCC) and University of Maryland Baltimore (UMB). Samples were collected with written informed consent and according to their local Institutional Review Board (IRB) approval at each institution. In addition, healthy volunteer blood samples were collected in CellSave preservative tubes™ or K2EDTA vacutainers with subject signed informed consent and IRB approval by Western IRB. All blood samples were kept at room temperature before filtration (storage at 4 °C causes formation of microclots in whole blood that can clog the filters).Cell lines
Tumor cell lines used in this study were purchased from ATCC (Manassas, VA). These include MCF-7, MDA-MB-231 and SK-BR-3 human breast cancer cell lines and PANC-1 pancreas epithelioid carcinoma. All cell lines were grown in their cell line specified media containing fetal bovine serum (FBS) as recommended by ATCC. Cell lines were maintained in T-25 or T-75 flasks using prescribed cell culture conditions (5% CO2, 37 °C) with media changes every 3–4 days, with the exception of the MDA-MB-231 cell line, which were grown at 37 °C with no added CO2. When fixed cells were used, cells were harvested using a trypsin–EDTA solution (ATCC Manassas, VA), spun at 125 × g for 5 min in 10% serum containing media to neutralize the trypsin, resuspended in PBS containing 1–4% paraformaldehyde (PFA) and incubated for 20 min at room temperature. After incubation, cells were diluted in 10× volume of PBS, centrifuged and resuspended in fresh PBS. When live cells were used, cells were harvested on the same day using trypsin–EDTA, neutralized as described above and resuspended in their specified serum-free media and stored for no more than 1 hour at room temperature before being spiked into normal blood and isolated using CellSieve™ microfilters within 5 min. CellSieve™ microfilters have been previously described as a 10 μm thick modified SU-8 polymer film with an array patterned filter with 7 μm diameter pores.6,17Capture and culture of live tumor cells spiked into normal blood
To evaluate cell viability by filtration, we spiked ∼100–1000 live MCF-7, PANC-1 SKBR3, and MB231, cells, into normal blood samples collected in K2EDTA vacutainers. Immediately after spike in (<1 min), the blood was drawn through the filters at 5 ml min−1 using a low pressure filtration system17 contained within an enclosed device (Fig. 2). After filtration and wash with PBS, as previously described,6,17,25 filters with the captured cells were placed into 12-well plates containing their specified serum-containing culture media. After 1–3 weeks the cell colonies were imaged on the filters. In addition, viability and cytotoxicity of the assay was evaluated using Calcein AM and POPO-3 (Life Technologies) according to manufacturer's protocols (ESI Fig. 1†).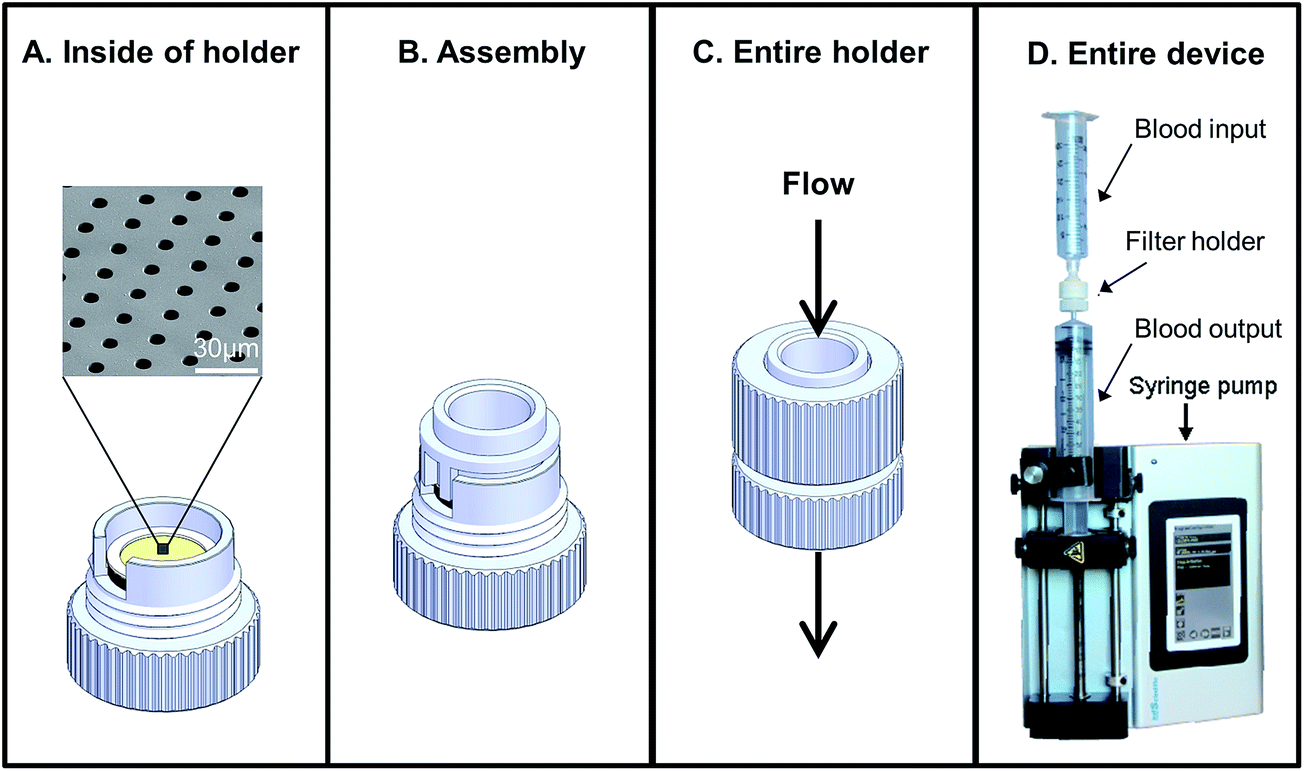 | ||
| Fig. 2 Flow through device with all-in-one reaction chamber. (A) The microfilter chip device consists of a holder and removable microfilter. (B) The device is designed with a reaction chamber which can be used run assays without then need to transfer the cells. (C and D) The entire device connects to sterile disposable syringes and a medical pump which allows uniform flow through the filter. | ||
Antibodies
A standard antibody mixture17 used in this study for staining epithelial cells consisted of FITC labeled-anti-cytokeratin 8, 18, 19; R-phycoerythrin (PE) labeled anti-EpCAM; and Cyanine5 labeled anti-CD45. Additional antibodies used include antibodies PE labeled anti-CD184 (CXCR4) antibody, and efluor 615 labeled anti-vimentin antibody.CTC staining procedures performed on CellSieve™ filters
Blood samples were filtered, fixed, permeabilized and washed, CTC identification by fluorescent enumeration was done as previously described.6,17 Filters were washed with PBS to remove unbound antibody, placed onto a microscope slide with Fluoromount-G/DAPI (Southern Biotech) and sealed with a glass cover slip. An Olympus BX54WI Fluorescent microscope with Carl Zeiss AxioCam was used to image the samples. Exposures were preset as 2–5 s (Cyanine5), 2 s (PE), 100–750 ms (FITC), and 10–50 ms (DAPI) for equal signal comparisons between cells. A Zen2011 Blue (Carl Zeiss) was used to process the images.Fluorescent in situ hybridization (FISH) analysis of cells captured by CellSieve™ filters
Following filtration and CTC immunostaining filters/cells can be probed using HER-2/CR-17 FISH probes performed as previously described25,26 with PathVysion HER-2 DNA Probe Kits, supplied by Abbott Molecular Inc. The identified CTCs x/y placement on the filter was marked on the filter substrate, and cell placement was recorded using Zen2011 Blue software (Carl Zeiss). Samples were demounted in a 2× SSC solution for 10 min and dried by air. The protease solution was added to each sample for 20 min in a 37 °C incubator. Slides were washed twice in 2× SSC for 5 min and were dried on a 45 °C warmer. Slides were placed in the denaturing solution at 72 °C for 5 min and were sequentially washed with 70%, 85%, and 100% ethanol for 1 min each, then dried on a 45 °C warmer, followed by the addition of 10 μl of probe to the slides, a coverslip was added, and sealed with rubber cement. The slide was incubated for 22 h in a 37 °C hybridization chamber. Coverslips were then removed in post-hybridization wash buffer at room temperature, washed with post-hybridization wash buffer at 72 °C for 2 min, rewashed in 2× SSC for 10 min, and dried at room temperature. Samples were mounted with Fluoromount-DAPI (Southern Biotech) and imaged on an Olympus BX54WI Fluorescent microscope with a Carl Zeiss AxioCam. Images were overlaid using Zen2011 Blue software (Carl Zeiss) as described.25H&E staining of cells on CellSieve™ microfilters
Following filtration and CTC immunostaining, cells/filters were further characterized with Hematoxylin and Eosin Y (Sigma), as previously described.26 After CTCs were imaged, filters were demounted in PBS and washed in PBS for 15 minutes. Filters were transferred to a microscope slide and filters were placed into a hematoxylin solution (Sigma) for 2 minutes. The slides were then rinsed 3 times in DI water then placed into an Eosin Y solution (Sigma) for 2 minutes, followed by briefly dipping in DI water until the desired color was achieved. Pre-imaged CTCs were then reimaged under white light.Isolating individual cells from CellSieve™ filters using micropipettes
Following filtration from blood, captured cells/filters were immediately incubated with permeabilization buffer for 15 minutes and then incubated with the CTC stain solution for 1 hour. Filters were washed with 5 ml 1× PBS/0.1% Tween-20. All steps were done without drying the sample. Cells identified as EpCAM positive, FITC positive and CD45 negative were removed using a micropipette with capillary pipette tips (Thermo Fisher) under an inverted dissecting microscope.Scanning electron microscopy (SEM) of CellSieve™ captured cells
For SEM, following filtration and CTC immunostaining, cells/filters were analyzed by SEM. After CTCs were imaged, filters were demounted in PBS and washed in PBS for 15 minutes. A solution of 2.5% glutaraldehyde in PBS was placed onto the filter and placed at 4 °C for 1 hour. Following incubation, filters were washed in DI water at room temp for 30 minutes. By holding the filter down on a microscope slide with tweezers, the filters were sequentially dehydrated through graded percentage of increasing ethanol – 70%, 80%, 90% and 100% ethanol for 2 minutes per each solution. Samples were dried, positioned onto a sample pin stub, and placed in a desiccator until imaged using a Phenom ProX Desktop Scanning Electron Microscope (NanoScience Instruments).For TEM, following filtration and CTC immunostaining, cells/filters were sliced by microtome for imaging.27 After CTCs were imaged, filters were demounted and washed in PBS for 5 minutes. Filters were transferred to a Swinnex filter holder (Thermo Fisher) and then sequentially dehydrated in ethanol – 70%, 80%, 90% and 100% ethanol for 30 seconds per each solution. The filter holder, with filter, was placed into a 37 °C incubator and pre-warmed liquid PEG 1000 (Sigma) was added to the filter holder and incubated for 30 minutes.27 The filtered holder was placed at −80 °C freezer for 1 hour. The filter was removed from the filter holder and placed onto a microtome specimen holder. The PEG block was cut cold on a rotary microtome, at 5–10 μm slices, and slices were transferred to poly-L-lysine coated slides.
Photobleaching and restaining of cells captured on CellSieve™ filters
The filtered and immunostained CTCs were further subtyped using additional immunomarkers. CTCs x/y placement on the filter was marked on the filter substrate and the cell placement was recorded using Zen2011 Blue software (Carl Zeiss). Samples were then archived and placed in storage at 4 °C for ∼2 years. Samples were removed from storage and PE fluorescence was photobleached by exposure to the excitation fluorescence (565 nm) for ∼10 seconds. Samples were demounted and placed into a filter holder. Cells on filters were again permeabilized for 20 min at RT and restained using an antibody panel of CXCR4 and vimentin, in the PE channel and eflour 660 channel, respectively. Filters were washed, placed onto a microscope slide with Fluoromount-G/DAPI (Southern Biotech) and sealed with a glass cover slip. An Olympus BX54WI Fluorescent microscope with Carl Zeiss AxioCam was used to re-image all bleached CTC. Exposures were preset as 500 ms (efluor 660) and 2 s (PE), and 10–50 ms (DAPI) for equal signal comparisons between cells. A Zen2011 Blue (Carl Zeiss) was used to process the images.Capture and backwash recovery of tumor cells and CTCs from patient samples using CellSieve™ filters
Prior to cell elution (backwash) of cells captured by CellSieve™, the filters were first blocked with 100% FBS for ∼10 min at room temperature, to prevent non-specific cell adherence. Capture efficiencies and contamination rates were calculated by spiking a known number of MCF-7 cells spiked into CellSave collected blood. A syringe pump (KD scientific) was used to provide low pressure vacuum filtration in a controlled flow format preventing accidental dehydration of the filter. Filtration was performed using CellSieve™ filters enclosed in a specialized filter holder, reducing blood cell retention (Fig. 2). After filtration and wash, the used syringe was removed and a clean syringe with 10 ml PBS was placed onto the bottom of the filter holder. The filter holder was placed upside down over a 15 ml conical tube and the PBS was gently pushed through the filter by hand, ∼10 ml min−1 for evaluating capture efficiency (ESI Fig. 2†). The eluted cells were then re-isolated on a fresh filter mounted on a normal filter holder. Both filters were then counted for the presence of CTCs and contaminating blood cells using a Zen2011 Blue (Carl Zeiss) software to count the cells, to determine the capture efficiency and determine the total backwash efficiency.The backwash procedure was also used to recover CTCs captured from patient samples. Sixteen patient samples drawn into 2 duplicate CellSave tubes, one tube was tested for capture efficiency and the duplicate tube was tested for backwash release. The first patient blood tube sample was run and CTCs were counted according to the standard CellSieve™ microfiltration assay as described.6,17 In conjunction, the second patient blood tube sample was run in same manner, but filter captured cells were eluted from the filters using the backwash method. CTC and contamination counts were then compared between the 2 sets of filtered samples (ESI Fig. 3†).
Results and discussion
The CellSieve™ microfiltration assay isolates CTCs using size based separation and is applicable to numerous clinical assays when used in singleplex CTC assessment.17,25 Interestingly, CTCs isolated by this system were described as clinically correlated to the CellSearch® assay,6 and thus allows for more detailed analysis of a specific and prognostically valuable CTC population. In the present study we expand on these initial singleplex assays, describing the evaluation of multiple biological techniques on rare CTCs directly isolated from whole blood patient samples.Summary of experimental protocols used in this study
We first developed the filtration and multiplex processes using cancer cell lines spiked into whole peripheral blood (Fig. 1A). We then validated our filtration and multiplex assays on patient samples (Fig. 1B). Finally as a proof of principle concept, we filtered cell lines, cultured the cells on the filters and ran sequential multiplex analysis on the cultured cells (Fig. 1C).As in a real clinical setting there are two possible starting points for utilizing CTCs, (1) shown in green/purple, blood collected in CellSave tubes for direct clinical evaluation of cells, without culture (Fig. 1A and B); or (2) shown in red are the protocols used for cell lines spiked into whole blood collected in EDTA vacutainers for indirect clinical evaluation after the captured cells are cultured and expanded (Fig. 1C). This system could provide isolation, culture expansion, and/or clinical relevant testing of the CTCs (Fig. 1C) and lead to broad study of cancer in real time by providing a supply of cancer cells. Unfortunately, while numerous groups are optimizing the specific environmental components for CTC expansion in tissue culture,24,28,29 the exact factors needed for consistent CTC culture are not known. Therefore, in this study we focused solely on isolation and in situ studies of CTCs using a microfluidic platform, knowing that the CTC culture is a separate study.
Capture and viability of live tumor cells in whole blood
Many microfilters developed for CTC isolation are made of toxic, harsh materials which are not conducive, or not capable of efficient capture of cells while retaining cell viability during isolation.17,24,30–35 To examine the effects of filtration on cell viability and cell growth patterns, we spiked a breast cancer cell line (MCF-7), which grows in clustered domes, and a pancreatic cell line (PANC-1), which grows as a monolayer sheet. Cells were spiked into EDTA blood vacutainers, isolated by filtration, and grown in the filtration unit (Fig. 2). We found the cell lines adhered and grew on CellSieve™ filters in their preferred growth pattern, i.e. a domed clustered structure for MCF-7 (Fig. 3A) or a monolayer sheet as seen with PANC-1 cells (Fig. 3B). Both cell lines were grown for 7 days in culture, and stained with CTC antibody cocktail including DAPI. Using these model cell lines, our results show that cells spiked into blood can be isolated on and subsequently grown within a filter based microfluidic holder. Additionally, using cell lines with two different growth properties, monolayer and clustered island, it was possible to track cancer growth behavior, suggesting that the filter polymer does not negatively affect cell viability or growth properties of the cells (Fig. 3 and ESI Fig. 1†). | ||
| Fig. 3 Isolation, culture and expansion of cells isolated on CellSieve™. (A) Live MCF-7 cells spiked into vacutainers, isolated by filtration and grown on the filter for 2–3 weeks. The 3 dimensional clustering attributed to this cell line can be seen on the filter. (green = anti-cytokeratin, blue = DAPI) (B) live PANC-1 cells spiked into vacutainers, isolated by filtration and grown on the filter for 2–3 weeks. This cell line can be seen growing as a monolayer on the filter. (C) SKBR3 cancer cell line is spiked into blood collected by CellSieve™. The CTCs are identified using presence of anti-cytokeratin and anti-EpCAM, with absence of anti-CD45. After CTCs are counted the cells are subtyped by HER2 FISH. (D) Live SKBR3 cells spiked into vacutainers, isolated by filtration and grown on the filter for 2–3 weeks. The expanded colonies can be directly analyzed as a whole colony, or as individual cells, molecularly by HER2/CR17 FISH analysis. (E) After filtration, a single CTC can be identified and harvested using a micropipette. (F) Removal of a single cell for downstream analysis (i.e. whole genome amplification, mRNA analysis). (G) After filtration, cells can be identified with histopathological stains (e.g. H&E) for cytological analysis or (H) after H&E, external cell structures can be analyzed by SEM. | ||
Despite our attempts, we have been unable to grow captured CTCs from cancer patient blood samples using published methods.24,29,36,37 However, these studies have shown that most CTCs isolated from cancer patient blood samples, although they may be technically alive, are incapable of further proliferation, requiring customized culture media which as of yet this not been optimized.1–3,24,29,35,37 In previously published works, less than 10% of late stage cancer patients have cultural CTCs and these CTCs senesce rapidly.1–3,24,29,35,37 As we observed no inhibitory effect on cell growth on the filters, we can only theorize that a lack of optimized culture parameters for propagating CTCs from cancer patients is the likely issue. As such, once CTC culture is possible, additional studies should be performed using the optimized media.
Multiplex CTC assays with a single device
Sequential clinical assays can be performed on cells isolated from whole peripheral blood. The entire proof of concept of multiplex in situ assaying is shown in Fig. 3. After cells are isolated, they can be cultured (Fig. 3A and B), and identified by chromogenic or fluorescent stains (Fig. 3A–E) and then evaluated for other subtyping assays (Fig. 3E–H). In theory the entire process could include CTCs in blood isolated by filtration and then either grown in tissue culture media; and/or stained; and/or molecularly characterized by FISH; and/or removed for single cell analysis; and/or histopathologically characterized by H&E stain.After optimizing the isolation and analytical procedures using cell lines (Fig. 1A) we transitioned to CTCs isolated from patient samples (Fig. 1B). Sixteen breast and prostate cancer patient samples were filtered and stained for classical CTC markers. Since the CellSieve™ low shear stress has been shown to preserve fine intracellular structures for cytological analysis;6,25 we assessed the clinical ramifications of cytological subtyping. In patient derived CTCs, we observed cells in various stages of the cell cycle, including apoptosis, (Fig. 4A and B) and mitosis (Fig. 4C and D). These CTC subtypes have been show as clinically relevant as the presence of apoptotic CTCs are associated with better prognosis than the presence of mitotic CTCs.6,23
 | ||
| Fig. 4 Cytological analysis and subtyping of CTCs from patients (A) CTC from a breast cancer patient categorized as early apoptotic with punctate cytokeratin, and an nucleus that appears as malignant with an abnormal salt-and-pepper pattern. This CTC subtype is associated with a favorable outcome.6,19 (B) CTC from a breast cancer patient categorized as late apoptotic with punctate cytokeratin and a nucleus which also appears punctate, or blebbing. This CTC subtype is associated with a favorable outcome. (C and D) CTCs from 2 breast cancer patients in the final stages of division (i.e. telophase/cytokinesis). This CTC subtype is associated with poor prognosis. Scale = 30 μm box. | ||
In standard tissue biopsies, cancer cells are first identified and graded and then cells are subtyped using a multitude of additional biomarkers (i.e. Ki-67, ER, PR, HER2, vimentin, etc.). In most other liquid biopsy techniques, CTCs are identified, enumerated, and might be subtyped, for a total of 2–3 biomarkers.8,38 For CTCs to yield similar clinical information as tissue biopsies, multiple additional markers must be used for subtyping CTCs.5,11,23,25,26,28 To establish a multi-biomarker panel, we took advantage of the anti-CD45 antibody, which is Cyanine5 labeled and is negative in CTCs; and therefore is open to staining with a CTC reactive Cyanine5 fluorescent antibody. Additionally, the fluorophore R-phycoerythrin (PE) attached to the EpCAM marker is bright but it is not photostable and prone to bleaching. After imaging the EpCAM, we used the photo instability to bleach the PE fluorescence signal by illuminating the cells for ∼10 seconds at 560 nm, thus facilitating the use of a new PE-labeled antibody. For illustration purposes, after bleaching, the samples were re-stained and subtyped with an antibody mixture of CXC chemokine receptor type 4 (CXCR4)-PE and vimentin-efluor 660 (Fig. 5).
 | ||
| Fig. 5 Bleaching and restaining CTCs After identifying and imaging patient derived CTCs using epithelial cell markers (e.g. cytokeratin+, EpCAM+ and CD45−), the PE fluorescence from EPCAM was bleached, freeing the channel for an additional marker. The CD45 was negative, allowing the channel to remain open for an additional marker. The DAPI channel and the cytokeratin-FITC channel remained unchanged, and can be used to identify the CTCs after restaining. The sample was then restained with the mesenchymal marker vimentin with efluor 660 and a stromal regulation marker CXCR4 with PE. Scale = 72 μm box. | ||
The ability to continue with further molecular characterization of CTCs using additional profiling greatly enhances their clinical utility. Thus far, we have described the ability to isolate, identify, proteomically subtype, genomically subtype and finally archive patient derive CTCs. We accomplish this all from a single sample in a sequential multiplexing manner to probe each CTC as an individual cell, or as a population of cells, all in relation to real world clinical utility.
Capture and backwash of tumor cells spiked into whole blood using CellSieve™ microfilters
Previously we demonstrated that tumor cells spiked into whole blood can be efficiently captured (∼90%) by CellSieve™ filters.17 Since many analytical methods require CTCs to be in solution phase (i.e. mRNA, single cell WGA, etc.), we examined whether cancer cells could be released from the filter after capture. For proof of concept, we determined the release capability of the assay by first spiking a known number of fixed cancer cells into whole blood and capturing them by filtration, as described (Fig. 1A).17 The captured cells are not covalently fixed onto the CellSieve™ filter and can therefore be easily eluted from the filter by injecting buffer through the filter holder in a retrogressive manner. After capture and wash, the cells were eluted from the filter using a backwash procedure, which consist of pushing buffer through the filter outlet and collecting the eluted cells into a 15 ml conical tube (ESI Fig. 2†). The eluted cells were then applied to a second filter to calculate both the capture efficiency and release rate of the cancer cells.Using MCF-7 tumor cells we find that cell capture efficiency is ∼90%, consistent with our previous results,17 and 97% ± 2% of the MCF-7s were released from the first filter (Fig. 6). We then enumerated white blood cells (WBC) that were captured along with the tumor cells to determine the rates of contaminating WBCs. Using 24 hour old blood, collected in CellSave tubes, we find that that ∼30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 WBCs were captured along with the tumor cells on the second filter with a calculated release rate of ∼97% (Fig. 6). However, being that human blood contains ∼10 × 107 WBCs per sample39 this is a 10000-fold purification, or a >99.9% removal of the WBCs from the blood sample, while retaining 90% of the spiked cancer cells. As patient derived CTCs are subjected to shear stress in the circulation and are considered “fragile” we looked to test patient isolated CTCS.40 To examine whether this backwash procedure can be performed on CTCs from cancer patient blood samples we used duplicate 7.5 ml blood samples collected from 16 cancer patients. One 7.5 ml tube was used to capture and analyze CTCs using our standard staining protocol,17 while in parallel, a second 7.5 ml blood sample from the same patient was used to evaluate the capture and release rates. CTCs on both filters were then identified by fluorescent enumeration as previously described.17 By running an interassay comparative study we find a CTC correlation of R2 = 0.97, p < 0.001, between the two assays indicating high concordance. Additionally, by purifying patient derived CTC, this data suggests that the CellSieve™ material is not reactive with circulating cells and that CTCs can be isolated and resuspended for additional downstream experimentation.
000 WBCs were captured along with the tumor cells on the second filter with a calculated release rate of ∼97% (Fig. 6). However, being that human blood contains ∼10 × 107 WBCs per sample39 this is a 10000-fold purification, or a >99.9% removal of the WBCs from the blood sample, while retaining 90% of the spiked cancer cells. As patient derived CTCs are subjected to shear stress in the circulation and are considered “fragile” we looked to test patient isolated CTCS.40 To examine whether this backwash procedure can be performed on CTCs from cancer patient blood samples we used duplicate 7.5 ml blood samples collected from 16 cancer patients. One 7.5 ml tube was used to capture and analyze CTCs using our standard staining protocol,17 while in parallel, a second 7.5 ml blood sample from the same patient was used to evaluate the capture and release rates. CTCs on both filters were then identified by fluorescent enumeration as previously described.17 By running an interassay comparative study we find a CTC correlation of R2 = 0.97, p < 0.001, between the two assays indicating high concordance. Additionally, by purifying patient derived CTC, this data suggests that the CellSieve™ material is not reactive with circulating cells and that CTCs can be isolated and resuspended for additional downstream experimentation.
 | ||
| Fig. 6 CTC release efficiency from microfilters for downstream analysis. To determine the ability of the CellSieve™ filters to backwash cells off the filters. MCF-7 cells (∼50 cells) were spiked into 7.5 ml normal blood and filtered. Cells were then backwashed off the filters and counted, as was the number of both CTCs and WBCs remaining on the filter. Y-axis shows the percentage of cells captured on the second filters versus cells still remaining on the first filters. Black bar, MCF-7 cells backwashed off the first filter and captured on the second filter versus cells remaining on the first filter. Striped bar = normal WBCs backwashed off the first filter and captured on the second filter versus cells remaining on the first filter. White bar, 16 patient samples of CTCs processed as described for the MCF-7 cells. Striped bar, same 16 patient samples of WBCs backwashed off the filter versus remaining on the filter as described above. | ||
Conclusions
While many technologies have been developed to capture CTCs from cancer patients, the rarity and fragility of CTCs coupled with the lack of commercially available platform technologies have limited the broad scale CTC analysis.5,9,12 Additionally, most CTC isolation platforms, are not conducive to multiple downstream clinical assays (i.e. Blood smears do not retain viability, microfluidics are slow, and density gradients require additional purification, etc.).5,9,12,18,21,35 These facts make detailed clinical testing and replication of CTC analysis difficult. We have previously described the ability to isolate, identify and test clinical assays on single CTCs from patients using commercial microfilters. Here we describe the next step in a clinical system, a disposable device that can be used as an all in one multiplexing platform with commercially available components (Fig. 2). We used this device to show initial proof of principle. We first described an optimal CTC workflow (Fig. 1) which would allow: (1) CTC isolation, (2) CTC culture, (3) CTC identification and (4) multiplex subtyping on a molecular and proteomic level. Cancer cells spiked into blood samples showed that microfilters were both conducive to culture, and the filtration process did not appear to alter cell growth behavior (Fig. 3). We then stained the cultured cells with CTC identification markers (i.e. cytokeratin and EPCAM) followed by FISH analysis, H&E histopathological stains, single cell separation, and numerous additional cell-based analysis (Fig. 3). To further elaborate on the possible downstream processing possibilities, we tested cancer cell elution from the filter to provide CTCs in suspension as required (i.e. single cell WGA, mRNA analysis, pharmacokinetics, etc.), and successfully applied this procedure to cancer patient CTCs. We showed that patient derived CTCs easily release from the filters, and thus would be a simple method to rapidly and efficiently purify CTCs for assays requiring suspended cells (Fig. 6).The main advantage of tissue biopsies over liquid biopsies is the plethora of clinically validated proteomic and genomic subtyping assays that can be performed on the samples. Our proof of principle study demonstrates that precision microfilters are ideal lab-on-a-chip platforms for studying CTCs, applicable to downstream analytical and staining techniques. Although staining CTCs with basic epithelial and tumor specific cell markers can determine their tissue origin, expanded molecular analysis can reveal specific tumor expressed mutations/amplifications. We have illustrated the first fluidic platform which isolates CTCs applicable for analysis using standard tissue protocols. Histopathological cell characteristics, commonly applied to cytology samples, can be applied to these CTCs, e.g. apoptotic and mitotic events present in CTCs (Fig. 4).6,23 Considering that mitosis in tumor cells is used as a predictive factor to inform therapeutic decisions, the ability to analyze CTCs using a mitotic index is likely to similarly inform therapy decisions during the life time of treatment. Furthermore, we suggest that histopathological assessment of CTCs can be followed by sequential proteomic and genomic profiling of cells, allowing sequential testing in regards to predictive medicine.
Despite the harsh environment of whole blood, the shear stress of isolation and the fragility of CTCs, we showed that that it is possible to purify patient CTCs while retaining the clinically important cytological information. We used this cytological based assessment in the same manner as classical histopathology to determine prognostic significance.6,23 We then went further, showing the ability to additionally subtype patient CTCs using a number of biomarkers within the same device. Again, as a proof of principle we added the mesenchymal marker vimentin, and the motility marker CXCR4, to show that proteomic multiplexing of CTCs is possible. While cell line propagation was successful, our initial attempts to culture CTCs from patients did not succeed. However, being that no group has consistently cultured CTCs, likely do to the high mortality of CTCs,24,28,29 we did prove that filters can be used as a culture surface. We showed that cells lines grew equivalent to standard culture techniques, unaffected by the filters or the filtration process. This is in contrast to other CTC isolation technologies which do not culture directly within their systems, but require additional steps to remove cells prior to culture. Using a single platform for all applicable assays allows for a more streamlined all-in-one assay which includes isolation, culture, proteomic testing and genomic testing without cell loss. Here we describe that an all-in-one system is feasible and that our initial testing using clinical patient samples already allows for in depth subtyping on a histopathological basis, which has not been described previously. While these data clearly call for additional studies, we present a simple, commercially applicable method, to purify CTCs, identify the cells with numerous biomarkers, while retaining the ability to characterize the CTCs with a variety of downstream techniques.
Acknowledgements
We would like to thank all of the patients who contributed to this study. This work was supported by a Maryland TEDCO MTTCF award, grants R01-CA154624 from the National Cancer Institute, KG100240 from the Susan G. Komen Foundation, a grant from an Era of Hope Scholar award from the Department of Defense (BC100675) and Department of Defense Advanced Research Projects Agency (W911NF-14-C-0098).References
- T. P. Butler and P. M. Gullino, Cancer Res., 1975, 35, 512–516 CAS.
- Y. S. Chang, E. di Tomaso, D. M. McDonald, R. Jones, R. K. Jain and L. L. Munn, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14608–14613 CrossRef CAS PubMed.
- I. J. Fidler, J. Natl. Cancer Inst., 1970, 45, 773–782 CAS.
- N. Hanna and I. J. Fidler, J. Natl. Cancer Inst., 1980, 65, 801–809 CAS.
- S. A. Joosse, T. M. Gorges and K. Pantel, EMBO Mol. Med., 2015, 7, 1–11 CrossRef CAS PubMed.
- D. L. Adams, S. Stefansson, C. Haudenschild, S. S. Martin, M. Charpentier, S. Chumsri, M. Cristofanilli, C. M. Tang and R. K. Alpaugh, Cytometry, Part A, 2015, 87, 137–144 CrossRef PubMed.
- S. J. Cohen, C. J. Punt, N. Iannotti, B. H. Saidman, K. D. Sabbath, N. Y. Gabrail, J. Picus, M. Morse, E. Mitchell, M. C. Miller, G. V. Doyle, H. Tissing, L. W. Terstappen and N. J. Meropol, J. Clin. Oncol., 2008, 26, 3213–3221 CrossRef PubMed.
- M. Cristofanilli, G. T. Budd, M. J. Ellis, A. Stopeck, J. Matera, M. C. Miller, J. M. Reuben, G. V. Doyle, W. J. Allard, L. W. Terstappen and D. F. Hayes, New. Engl. J. Med., 2004, 351, 781–791 CrossRef CAS PubMed.
- P. Paterlini-Brechot and N. L. Benali, Cancer Lett., 2007, 253, 180–204 CrossRef CAS PubMed.
- W. J. Allard, J. Matera, M. C. Miller, M. Repollet, M. C. Connelly, C. Rao, A. G. Tibbe, J. W. Uhr and L. W. Terstappen, Clin. Cancer Res., 2004, 10, 6897–6904 CrossRef PubMed.
- P. G. Febbo, M. Ladanyi, K. D. Aldape, A. M. De Marzo, M. E. Hammond, D. F. Hayes, A. J. Iafrate, R. K. Kelley, G. Marcucci, S. Ogino, W. Pao, D. C. Sgroi and M. L. Birkeland, J. Natl. Compr. Cancer Network, 2011, 9(5), S1–S32 CAS.
- E. S. Lianidou and A. Markou, Clin. Chem. Lab. Med., 2011, 49, 1579–1590 CrossRef CAS PubMed.
- S. K. Arya, B. Lim and A. R. Rahman, Lab Chip, 2013, 13, 1995–2027 RSC.
- I. Cima, C. Wen Yee, F. S. Iliescu, W. M. Phyo, K. H. Lim, C. Iliescu and M. H. Tan, Biomicrofluidics, 2013, 7, 11810 CrossRef PubMed.
- Y. Dong, A. M. Skelley, K. D. Merdek, K. M. Sprott, C. Jiang, W. E. Pierceall, J. Lin, M. Stocum, W. P. Carney and D. A. Smirnov, J. Mol. Diagn., 2013, 15, 149–157 CrossRef PubMed.
- C. Jin, S. M. McFaul, S. P. Duffy, X. Deng, P. Tavassoli, P. C. Black and H. Ma, Lab Chip, 2014, 14, 32–44 RSC.
- D. L. Adams, P. Zhu, O. V. Makarova, S. S. Martin, M. Charpentier, S. Chumsri, S. Li, P. Amstutz and C. M. Tang, RSC Adv., 2014, 9, 4334–4342 RSC.
- K. Pachmann, O. Camara, A. Kavallaris, S. Krauspe, N. Malarski, M. Gajda, T. Kroll, C. Jorke, U. Hammer, A. Altendorf-Hofmann, C. Rabenstein, U. Pachmann, I. Runnebaum and K. Hoffken, J. Clin. Oncol., 2008, 26, 1208–1215 CrossRef PubMed.
- S. Zheng, H. Lin, J. Q. Liu, M. Balic, R. Datar, R. J. Cote and Y. C. Tai, J. Chromatogr., 2007, 1162, 154–161 CrossRef CAS PubMed.
- E. A. Punnoose, S. K. Atwal, J. M. Spoerke, H. Savage, A. Pandita, R. F. Yeh, A. Pirzkall, B. M. Fine, L. C. Amler, D. S. Chen and M. R. Lackner, PLoS One, 2010, 5, e12517 Search PubMed.
- V. Gupta, I. Jafferji, M. Garza, V. O. Melnikova, D. K. Hasegawa, R. Pethig and D. W. Davis, Biomicrofluidics, 2012, 6, 24133 Search PubMed.
- M. Yu, S. Stott, M. Toner, S. Maheswaran and D. A. Haber, J. Cell Biol., 2011, 192, 373–382 CrossRef CAS PubMed.
- D. Adams, S. Martin, S. Chumsri, M. Charpentier, R. Alpaugh, M. Cristofanilli, C. Tang and C. Haudenschild, J. Clin. Oncol., 2015, 33, 11029 CrossRef PubMed.
- K. Kolostova, M. Broul, J. Schraml, M. Cegan, R. Matkowski, M. Fiutowski and V. Bobek, Anticancer Res., 2014, 34, 3641–3646 Search PubMed.
- D. L. Adams, S. S. Martin, R. K. Alpaugh, M. Charpentier, S. Tsai, R. C. Bergan, I. M. Ogden, W. Catalona, S. Chumsri, C. M. Tang and M. Cristofanilli, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3514–3519 CrossRef CAS PubMed.
- D. Adams, P. Zhu, O. Makarova, S. Li, P. Amstutz and C. Tang, Cancer Res., 2012, 72, 2395 CrossRef PubMed.
- A. Mendez-Vilas and A. Diaz, Microscopy: Science, technology, applications and education, 2010, 2, 907–913 Search PubMed.
- C. L. Hodgkinson, C. J. Morrow, Y. Li, R. L. Metcalf, D. G. Rothwell, F. Trapani, R. Polanski, D. J. Burt, K. L. Simpson, K. Morris, S. D. Pepper, D. Nonaka, A. Greystoke, P. Kelly, B. Bola, M. G. Krebs, J. Antonello, M. Ayub, S. Faulkner, L. Priest, L. Carter, C. Tate, C. J. Miller, F. Blackhall, G. Brady and C. Dive, Nat. Med., 2014, 20, 897–903 CrossRef CAS PubMed.
- L. Zhang, L. D. Ridgway, M. D. Wetzel, J. Ngo, W. Yin, D. Kumar, J. C. Goodman, M. D. Groves and D. Marchetti, Sci. Transl. Med., 2013, 5, 180ra148 Search PubMed.
- F. A. Coumans, G. van Dalum, M. Beck and L. W. Terstappen, PloS One, 2013, 8, e61770 CAS.
- F. Farace, C. Massard, N. Vimond, F. Drusch, N. Jacques, F. Billiot, A. Laplanche, A. Chauchereau, L. Lacroix, D. Planchard, S. Le Moulec, F. Andre, K. Fizazi, J. C. Soria and P. Vielh, Br. J. Cancer, 2011, 105, 847–853 CrossRef CAS PubMed.
- M. Hosokawa, T. Hayata, Y. Fukuda, A. Arakaki, T. Yoshino, T. Tanaka and T. Matsunaga, Anal. Chem., 2010, 82, 6629–6635 CrossRef CAS PubMed.
- L. S. Lim, M. Hu, M. C. Huang, W. C. Cheong, A. T. Gan, X. L. Looi, S. M. Leong, E. S. Koay and M. H. Li, Lab Chip, 2012, 12(21), 4388–4396 RSC.
- G. Vona, A. Sabile, M. Louha, V. Sitruk, S. Romana, K. Schutze, F. Capron, D. Franco, M. Pazzagli, M. Vekemans, B. Lacour, C. Brechot and P. Paterlini-Brechot, Am. J. Pathol., 2000, 156, 57–63 CrossRef CAS PubMed.
- S. Zheng, H. K. Lin, B. Lu, A. Williams, R. Datar, R. J. Cote and Y. C. Tai, Biomed. Microdevices, 2011, 13, 203–213 CrossRef PubMed.
- R. A. Harouaka, M. D. Zhou, Y. T. Yeh, W. J. Khan, A. Das, X. Liu, C. C. Christ, D. T. Dicker, T. S. Baney, J. T. Kaifi, C. P. Belani, C. I. Truica, W. S. El-Deiry, J. P. Allerton and S. Y. Zheng, Clin. Chem., 2014, 60, 323–333 CAS.
- X. Liu, V. Ory, S. Chapman, H. Yuan, C. Albanese, B. Kallakury, O. A. Timofeeva, C. Nealon, A. Dakic, V. Simic, B. R. Haddad, J. S. Rhim, A. Dritschilo, A. Riegel, A. McBride and R. Schlegel, Am. J. Pathol., 2012, 180, 599–607 CrossRef CAS PubMed.
- J. B. Smerage, W. E. Barlow, G. N. Hortobagyi, E. P. Winer, B. Leyland-Jones, G. Srkalovic, S. Tejwani, A. F. Schott, M. A. O'Rourke, D. L. Lew, G. V. Doyle, J. R. Gralow, R. B. Livingston and D. F. Hayes, J. Clin. Oncol., 2014, 32, 3483–3489 CrossRef CAS PubMed.
- J. G. Hollowell, O. W. van Assendelft, E. W. Gunter, B. G. Lewis, M. Najjar and C. Pfeiffer, C. Centers for Disease and N. C. F. H. S. Prevention, Vital and health statistics, Series 11, Data from the national health survey, 2005, pp. 1–156 Search PubMed.
- L. M. Flores, D. W. Kindelberger, A. H. Ligon, M. Capelletti, M. Fiorentino, M. Loda, E. S. Cibas, P. A. Janne and I. E. Krop, Br. J. Cancer, 2010, 102, 1495–1502 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21524b |
| This journal is © The Royal Society of Chemistry 2016 |