
In situ ion-exchange synthesis of SnS2/g-C3N4 nanosheets heterojunction for enhancing photocatalytic activity†
Abstract
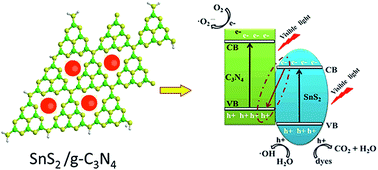
In this paper, free standing graphitic carbon nitride (g-C3N4) nanosheets have been synthesized by mixed solvents (water/IPA = 2/1) liquid phase exfoliation, and a series of SnS2/g-C3N4 heterojunctions with different contents of SnS2 have been prepared via a simple ion-exchange process. Exfoliated g-C3N4 presents a two-dimension sheet-like structure with the thickness of 2.8 nm and small SnS2 nanoparticles with diameter of 5–10 nm are well anchored on the surface of g-C3N4 nanosheets, which was proved by transmission electron microscopy (TEM) and atomic force microscope (AFM). The 4.0-SnS2/g-C3N4 sample shows the highest photocurrent density of 13.66 μA cm−2 at 0.8 V, which is about 1.5 time of the g-C3N4 nanosheets and 2 times of the bulk g-C3N4, respectively. Photocatalytic measurement also demonstrated that constructing heterojunction of SnS2/g-C3N4 can improve the photocatalytic efficiency as compared to pure g-C3N4 and g-C3N4 nanosheets. The highly effective photoelectrochemical and photocatalytic activities of SnS2/g-C3N4 heterojunctions are attributed to the efficient separation of photogenerated hole–electron pairs. This work may provide a novel concept for the rational design of high performance g-C3N4-based photocatalysts.
 Please wait while we load your content...
Please wait while we load your content...

