
In situ solution polymerization for preparation of MDI-modified graphene/hyperbranched poly(ether imide) nanocomposites and their properties
Quantao Li†
ab,
Wenqiu Chen†ab,
Wei Yanab,
Quanyuan Zhangab,
Changfeng Yi*ab,
Xianbao Wangab and
Zushun Xu*ab
aHubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Wuhan, 430062, China. E-mail: zushunxu@hubu.edu.cn; changfengyi@hubu.edu.cn; Fax: +86 27 88665610; Tel: +86 27 88661897
bMinistry-of-Education Key Laboratory for the Green Preparation and Application of Functional Materials, Hubei University, Wuhan 430062, China
First published on 8th December 2015
Abstract
Firstly, fully exfoliated graphene oxide (GO) colloidal dispersion in N-methyl-2-pyrrolidone (NMP) with high concentration is obtained by a solvent-exchange method and further used to prepare superior GO-MDI with free isocyanato groups by chemical modification. Then, the yielded GO-MDI is employed to prepare two kinds of MDI-modified graphene/hyperbranched poly(ether imide) (GE-MDI/HBPEI) nanocomposites via in situ random solution co-polycondensation or crosslinking reaction, followed by synchronous thermal imidization and reduction. The chemical modification of GO endows GO-MDI with good solubility in organic solvents to prepare GE-MDI/HBPEI nanocomposites with high filler content. GO-MDI is further used as a multi-functional co-monomer or crosslinker to be introduced into the HBPEI backbone with full compatibility of the guest and host at the molecular level. Finally, the performance tests show that the heat resistance, thermal stability, mechanical strength and modulus, and gas barrier properties of the obtained two kinds of nanocomposites are significantly improved or enhanced compared with pure HBPEI, and the impacts become more and more significant with the increase of GO-MDI content, but their mechanical toughness show trends of increase at first then decrease with the increase of GO-MDI content. Comparisons also show that at the same GO-MDI content, the heat resistance, thermal stability, mechanical strength and modulus of the nanocomposites obtained by in situ random solution co-polycondensation are all superior to those obtained by in situ random solution crosslinking reaction, except the mechanical toughness and gas barrier properties of the former are less than the latter. This effective approach provides a possibility for enriching and developing high performance PEI-based composites with various forms of GE for advanced engineering or functional materials.
1 Introduction
As one of the most important super thermoplastic engineering materials, poly(ether imide) (PEI) not only inherits the excellent mechanical, thermal, optical and electronic properties of aromatic polyimide (PI), but also solves the processing problem of PI resulted from the poor solubility, ultrahigh softening or melting temperature. So compared with PI, PEI has far more extensive applications in high-tech fields such as microelectronics, optoelectronics and aerospace, etc.1–3Hyperbranched polymer (HP) has attracted an increasing attention because of its unique structural characteristic, outstanding physical and chemical properties and potential application, especially the lots of terminal groups which are conducive to the subsequent functionization.4,5 Recently, in addition to keeping the superior high-temperature stabilities, high strength and modulus of PIs, various introduction of hyperbranched structure into PIs also endow their products with good organo-solubilities, low solution viscosity and amorphism.6–11 But some performance of the hyperbranched polyimides (HBPIs) which have been reported, leaves more or less to be desired, due to the special features of the molecular structure.12,13
As is known to all, various organic or in organic hybrid or composite materials based on PIs are also found to be one of the most simple and effective approaches for promoting the comprehensive performance or developing new functions.14–16 Graphene (GE) has been proved to be one of the most effective enhanced additives for linear PI-based composites, but the improvements is extremely limited due to the compatibility problem with PI.17 The individual or few properties of GE/PI nanocomposites which are derived from the precursor of GE (graphene oxide, GO) or its chemical modified product (CMG) with heavy oxygen-containing decoration,18 are very obviously promoted or improved for the good compatibility of the matrix and additive.19–24 Nevertheless, the preparation method of the GE/PI nanocomposites that is a simple solution mixing of GO or CMG and poly(amic acid) (precursor of PI, PAA), still faces some problems. One is the weak interaction between GO (or CMG) and the PI matrix, and the other is the decomposition of the labile oxygen-containing groups, which also negatively affects the interface interaction.19–22,25–28 Though the interfacial interaction problem of GE/PI nanocomposites could be settled by the incorporation of modified GO with various free reactive groups and PI backbones with strong covalent bonding between the host and guest,29–31 but the practical improvements of the nanocomposites are not as good as expected.32,33 For one thing, it attributed to the structural defect problem associated with the pyrolysis of GO in the thermal treatment.34 For another, GO can not be exfoliated readily to yield single-layer and dispersed uniformly or equably in most organic solvents with a GO content equal to or higher than 0.5 mg mL−1 in general.35–37
In order to improve the interfacial interaction of GE/PI nanocomposites, Zhang and co-workers38 reported that excess MDI was grafted onto the surface of GO nanosheet via the addition reaction between hydroxyl or carboxyl group and isocyanato group, and the yielded GO-MDI was further used to prepare functionalized GE/PI nanocomposites with highly enhanced mechanical and thermal properties by the reaction of the free isocyanato group of GO-MDI with the carboxyl group of PAA. Previously, GO was dispersed in the organic solvent DMF in advance by mechanical stirring and ultrasonic dispersing in DMF. However, GO cannot be exfoliated readily to yield single layer in most organic solvents even though an ultra-low solid content as mentioned above, but it can be easily exfoliated in water to yield GO colloidal dispersion with single layer and high concentration for the heavily decorated hydrophilic groups of GO. Recently, Liff39,40 and Wang41 obtained a high concentration of single layer dispersed colloidal dispersion of GO by solvent-exchange method. GO was dispersed in deionized water firstly, then the corresponding organic solvent was added into the aqueous solution, and water was removed by vacuum distillation at last.
As far as we know, almost all of the GO/PI or GE/PI composites reported focused on linear polymers, and little involved HBPIs as the matrix resins.
Based on the aforementioned considerations in the study, completely exfoliated GO colloidal dispersion in NMP was firstly obtained by solvent-exchange method, and then used to reacted with excess 4,4′-diphenylmethane diisocyanate (MDI) to afford MDI-modified GO (GO-MDI) with free isocyanato groups. Hereafter, combining with the experiences and conclusions of the novel hyperbranched poly(ether imide)s (HBPEIs) derived from the novel BB′2-type triamine 2,4,6-tris[4-(4-aminophenoxy)phenyl] pyridine (TAPPP) with symmetrical triaryl-substituted pyridine structure reported before,42 a series of co-polycondensed GE-MDI/HBPEI nanocomposites ((GE-MDI)-co-HBPEI) with various contents of GO-MDI, were prepared via in situ random solution co-polycondensation of GO-MDI with the novel triamine TAPPP, commercial bisphenoxyphenyl diamine and bisphenol A dianhydride, as well as subsequent synchronous thermal imidization and reduction. Meanwhile as a comparison, series of crosslinked GE-MDI/HBPEI nanocomposites ((GE-MDI)-g-HBPEI) with various contents of GO-MDI, were also obtained by in situ solution crosslinking reaction between the free isocyanato group of GO-MDI and the free terminal anhydride group of the pre-prepared precursor of the above HBPEI, and the same subsequent process. GO-MDI obtained by solvent-exchange method and further chemical modification is expected to not only increase its solubility in organic solvent with high GO-MDI content, but also is used as a multi-functional co-monomer or crosslinker to be introduced into the HBPEI backbone with a full compatibility of the guest and host in molecular-level. The chemical structure, composition and micromorphology, as well as the related properties of the GO-MDI were characterized by some means, and further comparative studies on the chemical and physical structure, thermal, mechanical and gas barrier properties of the obtained two kinds of GE-MDI/HBPEI nanocomposites were designedly demonstrated. Attempts were also made to correlate the micromorphology of the nanocomposites to the corresponding properties.
2 Experimental
2.1 Materials
Graphene oxide (GO) was obtained by a modified Hummer method as demonstrated.43,44 2,4,6-Tris[4-(4-aminophenoxy)phenyl]pyridine (TAPPP) was synthesized as our previous work.42 4,4′-Diphenylmethane diisocyanate (MDI, 99.9%) was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan) and used as received. 4,4′-Diaminodiphenyl ether (ODA, 98%) was obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China) and recrystallized from ethanol before use. 2,2-Bis[4-(3,4-dicarboxyphenoxy)phenyl]propane dianhydride (BPADA) was purchased from Shanghai Research Institute of Synthetic Resins (Shanghai, China) and recrystallized from acetic anhydride followed by drying in vacuum at 120 °C for 12 h prior to use. N-Methyl-2-pyrrolidone (NMP) and dichloromethane were distilled in the presence of calcium hydride under reduced pressure and stored over 4 Å molecular sieves. Other commercially available reagent grade chemicals were used without further purification.2.2 Preparation of MDI-modified graphene oxide (GO-MDI) solution in NMP
As shown in Scheme 1, firstly, GO (0.5 g) was exfoliated in 200 mL deionized water with 1 h ultrasonication. 100 mL NMP was then added, followed by stirring for 1 h and ultrasonication for another 0.5 h. The resulting mixture was distilled under reduced pressure to remove water until a portion of NMP was distilled out, followed by 0.5 h centrifugation at 3000 rpm. The concentration of GO in the supernatant was determined by weighing the sediments after dried.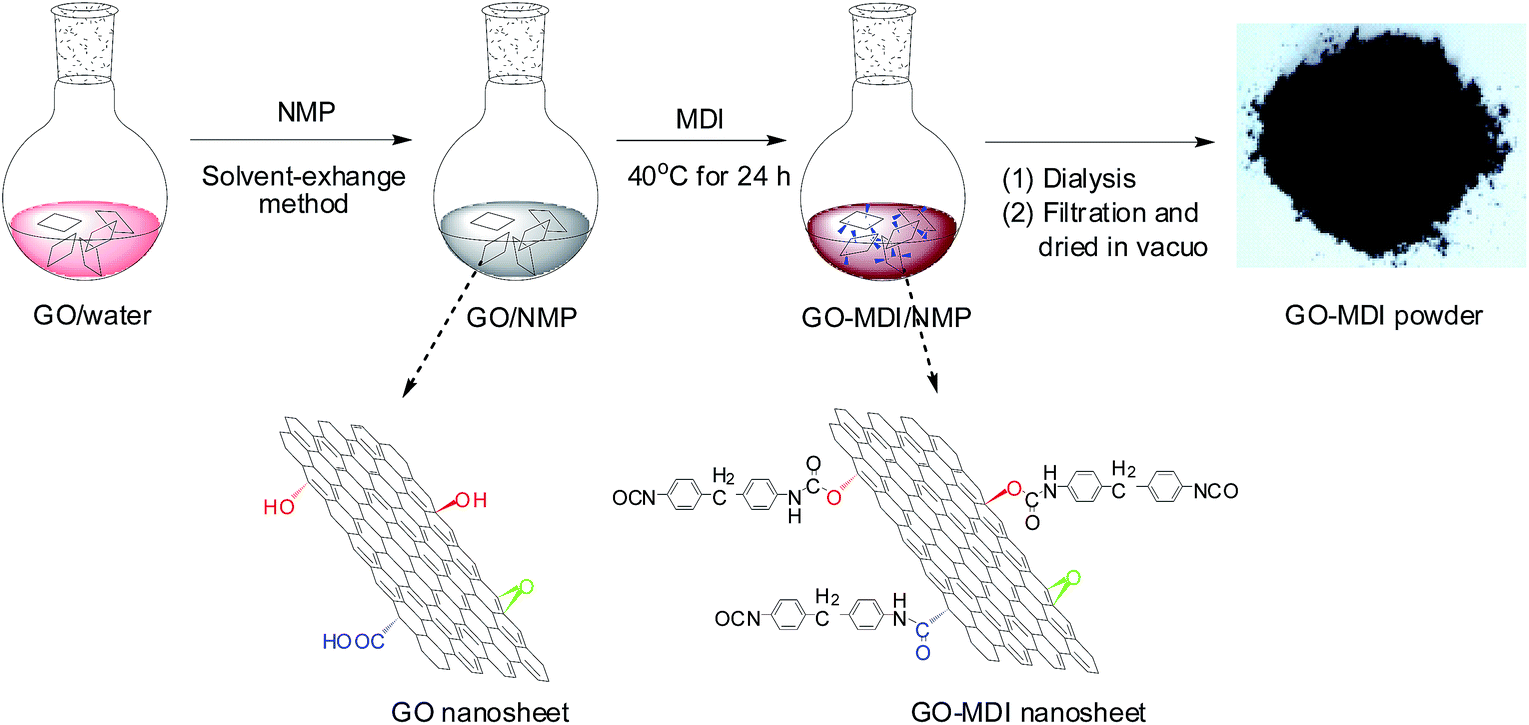 | ||
| Scheme 1 Synthetic process of GO-MDI. | ||
Hereafter, 3.0 g MDI was added into the as-prepared solution and stirred at 40 °C for 24 h under N2. The solution mixture was subjected to dialysis in dehydrated dichloromethane for 7 days to remove the residual MDI thoroughly. The modified GO with MDI (GO-MDI) was obtained after being filtered, dried at 80 °C in vacuo and grinded.
Finally, the obtained GO-MDI powder was dispersed in distilled NMP under vigorous stirring and then exfoliated by ultrasonication treatment to afford a GO-MDI colloidal solution with a concentration of 5 mg mL−1.
2.3 Preparation of GE-MDI/HBPEI nanocomposites
The GE-MDI/HBPEI nanocomposites were synthesized by the following procedures as shown in Scheme 2(a) and (b). A representative polymerization of GE-MDI/HBPEI nanocomposites (marked as (GE-MDI)-co-HBPEI) with 0.10 wt% GO-MDI is as follows: firstly, 0.718 g (1.38 mmol) BPADA, 0.22 mL of the as-prepared GO-MDI colloidal solution in NMP, and 9.46 mL NMP were placed in a completely dried 100 mL three-necked round-bottom flask. After being ultrasonicated for 1 h under N2, the mixture was conducted with stirring at 80 °C for another 2 h. Meanwhile, 0.200 g (1 mmol) ODA and 0.157 g (0.25 mmol) TAPPP were dissolved in 5 mL NMP in another 100 mL thoroughly dried three-necked round-bottom flask under N2 flow. This solution was then added dropwise to the above standby mixture, through a syringe with a duration of 1 h under N2 flow and magnetic stirring at 40 °C. The reaction mixture was further conducted for 12 h at 40 °C to afford the nanocomposite precursor solution. The nanocomposite film (about 0.10 ± 0.02 mm) was obtained by casting the precursor solution onto a leveled clean glass plate after filtered through a 400 mesh sieve. The casting procedure was conducted with a thermal treatment according to the following steps: 100 °C for 8 h, 150 °C, 200 °C, 250 °C and 300 °C each for 1 h in nitrogen atmosphere. The other (GE-MDI)-co-HBPEI nanocomposites (about 0.10 ± 0.02 mm) with 0.25 wt%, 0.50 wt%, 0.75 wt% and 1.0 wt% of GO-MDI were obtained via the above mentioned steps by adding a right amount of as-prepared GO-MDI dispersion respectively. Furthermore, the total volume of NMP derived from GO-MDI/NMP colloidal solution and NMP added alone was kept 15 mL. | ||
| Scheme 2 Synthetic process of (a) (GE-MDI)-co-HBPEI and (b) (GE-MDI)-g-HBPEI nanocomposites. | ||
For comparison, the same charging amount but different charging order was conducted to investigate the effects of the obtained nanocomposites on the properties. Firstly, a mixture of ODA and TAPPP was dropped into BPADA solution to afford a precursor of anhydride-terminated hyperbranched poly(amic acid) (AD-HBPAA) as a standby. Then appropriate amount of the as-prepared GO-MDI colloidal solution was added to the above precursor solution and conducted for some time. The other procedures were the same as mentioned above. The resulting product with 0 wt% of GO-MDI was actually the pure HBPEI, and others were marked as (GE-MDI)-g-HBPEI nanocomposites with right content of GO-MDI.
2.4 Measurements
Fourier transform infrared (FTIR) spectroscopy was recorded on a Nicolet iS50 spectrometer (Thermo Fisher Scientific, USA) by transmittance mode or surface reflection mode. Scanning electron microscopy (SEM) and energy dispersive X-ray (EDX) spectroscopy were performed on JSM-6510LV (JEOL, Japan). Atomic force microscopy (AFM) was carried out by using an SOLVER NANO SPM from NT-MDT. X-ray photoelectron spectroscopy (XPS) analysis was carried out on a SPECS system (Germany) with a PHOIBOS 150 analyser, and an MCD-9 detector. Raman spectrum was measured on a Lab RAM HR 800 UV (HORIBA Jobin Yvon, France) multi-channel confocal microspectrometer with 633 nm laser excitation. X-ray diffraction (XRD) pattern was conducted on a D8-Advance diffractometer (Bruker, Germany) with a CuKα radiation source (λ = 0.15418 nm). The dynamic mechanical thermal analysis (DMTA) was performed with a Q800 DMA (TA Instruments, USA) at 1 Hz, in the temperature range from 50 °C to about 400 °C, at a heating rate of 5 °C min−1 in air. Differential scanning calorimetry (DSC) was performed using a Q2000 differential scanning calorimeter (TA Instruments, USA) from 100 °C to 400 °C under N2 flow at a heating rate of 10 °C min−1. Thermogravimetric analysis (TGA) was carried out with a SII Diamond TG/DTA (Perkin-Elmer, USA) from room temperature to 800 °C at a heating rate of 10 °C min−1 under both air and N2 atmosphere. The mechanical property was measured on a CMT4104 Electromechanical Universal Testing Machine (Shenzhen SANS Testing Machine Co., Ltd., China) with 50 × 5 mm2 films at a load of 100 N and a rate of 2 mm min−1. The gas barrier property of with the size of 7.069 cm2 was measured using a VAC-V2 gas permeability tester (Labthink Instruments Co., Ltd., China) with reference to GB 1038 in N2 at 23 °C.3 Results and discussion
3.1 Preparation of MDI-modified graphene oxide (GO-MDI)
As reported,39–41 single layer dispersed and high concentration GO colloidal dispersion in NMP was prepared by solvent-exchange method firstly, and then it was modified with excess MDI to afford GO-MDI with free isocyanato groups on the surface. This ensures to improve the organic dispersibility of GO-MDI, and the subsequent feasible employment to prepare GE-MDI/HBPEI nanocomposites via in situ solution polymerization. The detailed synthetic process of GO-MDI is shown in Scheme 1.The dispersibility of GO-MDI in appropriate organic solvent is crucial to obtain homogeneous polymer nanocomposites. Fig. 1(a) compares the re-dispersions of GO-MDI powder in water, NMP, chloroform and benzene after ultrasonication and standing for 24 h. It is clearly shown that GO-MDI powder can be re-dispersed homogeneous in NMP with different contents, but it precipitates in water to the bottom of vial, or aggregates around the interface of water and chloroform or benzene, indicating that the modified product changes from hydrophilic to hydrophobic. Fig. 1(b) is the inverted photos of re-dispersions of GO-MDI in NMP with various contents. It is found that the dark brown re-dispersions of GO-MDI in NMP, even with the content as high as 5 mg mL−1, contain no visible precipitate and are stable for 24 h. Fig. 1(c) shows that there is obvious Tyndall phenomenon of re-dispersions of GO-MDI in NMP at 2 mg mL−1 by shining a laser light, which indicates that GO-MDI is actually colloidal dispersed in NMP. The well dispersibility of GO-MDI in NMP makes it an ideal candidate for advanced nanocomposites in the further solution polymerization.
 | ||
| Fig. 1 (a) Photos of GO-MDI redispersing in water, NMP, chloroform and benzene after ultrasonication and standing for 24 h; (b) the inverted photos of redispersions of GO-MDI in NMP with various contents after standing for 24 h; (c) the Tyndall phenomenon of redispersions of GO-MDI in NMP at 2 mg mL−1 by shining a laser light. | ||
The micromorphology, structure and elementary composition of GO and GO-MDI are displayed in Fig. 2. As SEM images shown in Fig. 2(a) and (b), GO aggregates randomly with obvious ultrathin, crumpled and unconsolidated morphology,45,46 and GO-MDI shows the same thin and silk-like structure, but little more thick and dense. Fig. 2(c) is the typical AFM images and corresponding height profile of GO-MDI nanosheet, which is dispersed in NMP with concentration of 0.01 mg mL−1. It could be seen that the average height of GO-MDI nanosheet deposited on silicon surface was about 1.06 nm, which is a little higher than that of GO nanosheet, for the local buckling or bulge of GO-MDI nanosheet.38 This change is because that the grafted MDI unites were inserted into the GO nanosheets and increased the distance between the layers. The EDX spectra of GO and GO-MDI obtained by the SEM images (Fig. 2(a) and (b)) area are showed in Fig. 2(d). After modified with MDI, about 18.84 wt% of nitrogen is detected, and the quality percentage of carbon increases from 68.57 wt% to 71.64 wt%, while the quality percentage of oxygen decreases from 31.43 wt% to 9.53 wt%, compared with GO. This is an indirect argument that MDI is grafted onto GO nanosheets, and the residual oxygen-containing groups on GO-MDI nanosheets are partly reduced under solvothermal effect when the modification is conducted in NMP.47,48 As shown in Fig. 2(e), natural graphite (NG) shows a sharp diffraction peak at 2θ = 26.2°, corresponding to the graphitic interlayer 002 with a spacing (d) of 0.34 nm, while GO exhibits a broad diffraction peak at 2θ = 10.7° (d = 0.83 nm).20 After modified by MDI, a smaller angle at 2θ = 9.3° (d = 0.95 nm) is observed. The increase of interlayer distance of GO-MDI indicates that MDI is successfully grafted onto the surface of GO nanosheets. Meanwhile, a much more broadening peak appears around 2θ = 20.0° (0.44 nm), closing to the graphitic interlayer spacing of NG (0.34 nm). It attributes to that the grafting reaction of MDI and GO is locally occurred, because the oxygen-containing groups are mainly distributed on the basal planes or edges of GO nanosheets, as well as the oxygen-containing groups on GO nanosheet are partly reacted with the isocyanato groups of MDI.
 | ||
| Fig. 2 SEM images of (a) GO and (b) GO-MDI, (c) typical AFM tapping-mode image and thickness of GO-MDI nanosheet, (d) EDX spectra and (e) XRD patterns of GO and GO-MDI. | ||
The detailed modification mechanism for MDI grafting onto GO nanosheet is analyzed by XPS. As shown in Fig. 3(a), compared with GO, the XPS spectrum of GO-MDI shows an obvious appearance of N1s peak (around 400 eV) and a reduction of the intensity of O1s peak (around 535 eV), which proves that the oxygen content is reduced after being modified by MDI. Minutely, C1s XPS spectrum of GO in Fig. 3(b) reveals the typical peaks for the carbon atom in various groups on the nanosheet surface, such as the non-oxygenated graphitic carbon (284.8 eV), the hydroxyl carbon (285.9 eV), the epoxy carbon (287.0 eV), the carbonyl carbon (287.8 eV) and the carboxylic acid carbon (288.8 eV), respectively.38 After modification, C1s XPS spectrum of GO-MDI in Fig. 3(c) is simulated with the carbon in C–N linkage (285.5 eV), the carbon in amido linkage (288.3 eV) and the carbon in urethane linkage (289.1 eV) in turn, except the non-oxygenated graphite carbon, the hydroxyl carbon and the epoxy carbon. These new chemical form carbons are derived from the reaction of isocyanato group and the hydroxyl or carbonyl group.38 Moreover, the N1s XPS spectrum of GO-MDI in Fig. 3(d) can be divided into three peaks. The main peak at 400.3 eV is attributed to the nitrogen in amido linkage and urethane linkage. The other two peaks at 399.2 eV and 401.6 eV, isocyanato group in turn, indicate that isocyanato groups are existed on the surface of GO-MDI nanosheets and some of them are hydrolyzed to amino groups.38 According to the concentration of nitrogen atom (Fig. 3(a)) and the fitting result of N1s (Fig. 3(d)), the mass percentage of nitrogen atom in the isocyanato group on the surface of GO-MDI nanosheet can be roughly estimated for 4.5% together with the hydrogen atom ignored.
 | ||
| Fig. 3 (a) The XPS survey of GO and GO-MDI; high-resolution C1s XPS spectra of (b) GO and (c) GO-MDI; (d) high-resolution N1s XPS spectrum of GO-MDI. | ||
The chemical structure change before or after the thermal treatment process of GO-MDI, which is employed to prepare the final nanocomposites, is investigated by FTIR and Raman spectra. As shown in Fig. 4(a), comparing with the characteristic absorption peaks of hydroxyl group (3400 cm−1), carbonyl group (1727 cm−1) and epoxy group (1063 cm−1) of GO, it is clearly found that new peaks at 2252 cm−1, 1597 cm−1, 1545 cm−1 and 1508 cm−1 appears in the FTIR spectrum of GO-MDI. They are assigned to isocyanato group (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO stretching vibration), amino group (N–H deformation vibration), amido linkage or urethane linkage (C–N stretching vibration) and the phenyl framework vibration in MDI moieties, respectively. Furthermore, the peak at 1702 cm−1 is the CO stretching vibration in amido linkage or urethane linkage. These proved that MDI is successfully grafted onto the GO nanosheet with free isocyanato groups. In addition, compared with GO-MDI, the peaks of GO-MDI treated at 300 °C (GO-MDI-300 °C), assigning to the amido linkage or urethane linkage (CO stretching vibration) and the phenyl framework of MDI moieties, are still observed with little weakening, and the peaks of other oxygen-containing groups are all weakened significantly. It indicates that the thermal treatment makes most of the residual oxygen-containing groups decompose,34 but no obvious changes for the MDI moieties and GEs framework.
CO stretching vibration), amino group (N–H deformation vibration), amido linkage or urethane linkage (C–N stretching vibration) and the phenyl framework vibration in MDI moieties, respectively. Furthermore, the peak at 1702 cm−1 is the CO stretching vibration in amido linkage or urethane linkage. These proved that MDI is successfully grafted onto the GO nanosheet with free isocyanato groups. In addition, compared with GO-MDI, the peaks of GO-MDI treated at 300 °C (GO-MDI-300 °C), assigning to the amido linkage or urethane linkage (CO stretching vibration) and the phenyl framework of MDI moieties, are still observed with little weakening, and the peaks of other oxygen-containing groups are all weakened significantly. It indicates that the thermal treatment makes most of the residual oxygen-containing groups decompose,34 but no obvious changes for the MDI moieties and GEs framework.
 | ||
| Fig. 4 (a) FTIR and (b) Raman spectra of GO, GO-MDI and GO-MDI-300 °C. | ||
The Raman spectra of GO and GO-MDI in Fig. 4(b) exhibits the significant D-band at 1341 ± 5 cm−1 and G-band at 1588 ± 2 cm−1 without significant shift, just varying from the relative intensity.31 As an index to appraise the relative defect level and the degree of chemical modification of GEs, the intensity ratio of the D-band and G-band (ID/IG) for GO-MDI and GO-MDI-300 °C slightly decrease to 0.94 and 0.97 respectively, compared with that of GO 1.00. The similar value of ID/IG indicates that little defects are introduced, and the basic structural characteristic of GEs nanosheet without excessive disorder is reserved during the MDI modification and further thermal treatment process. Furthermore, sharp 2D-bands are observed at 2670 ± 4 cm−1 both for GO and GO-MDI, indicating that they are probably aggregated with a few layers stacking structure.49 But it do not exist in the Raman spectrum of GO-MDI-300 °C, because the accumulation or arrangement of the interlayer becomes more closely derived from the pyrolysis of labile oxygen-containing moieties.34
TGA is a reliable method to evaluate the thermal stability of different structural forms of carbon materials as well as their modification effect. As shown in Fig. 5, the TGA curve of powdery GO shows about 8% weight loss before 150 °C, evidently due to the evaporation of the adsorbed water molecules. The most significant weight loss (about 25%) is observed between 150 °C and 250 °C, which is attributed to the loss of CO, CO2 and steam resulted from the pyrolysis of the labile oxygen-containing groups.34 Since then until 800 °C, there is a weight loss about 20 wt% caused by the pyrolysis of the oxygenated graphitic carbon skeleton of GO. The TGA curve of GO-MDI exhibits almost no weight loss before 150 °C, indicating that the residual water is removed completely. The main weight loss occurs in the temperature range of 150–250 °C (about 15%) and 250–600 °C (about 30%), because of the decomposition of the residual oxygen-containing groups and the decomposition of the grafted MDI moieties together with the oxygenated graphitic carbon skeleton of GO, respectively. The former weight loss is less than that of GO, indicating that some of the oxygen-containing species on GO-MDI are reduced by solvothermal effect in the modification process of GO. As a comparison, GO-MDI-300 °C shows little weight loss before 300 °C, for the residual oxygen-containing species are further reduced with few remaining, and about 25 wt% of the weight loss between 300–600 °C which is also attributed to the decomposition of the grafted MDI moieties and the oxygenated graphitic carbon skeleton of GO.
 | ||
| Fig. 5 TGA curves of GO, GO-MDI and GO-MDI-300 °C in N2. | ||
All the above results clearly indicate that MDI is mainly grafted onto GO nanosheet with free isocyanato group. Meanwhile, it also provides an effective way to prepare a homogeneous and stable suspension of modified GO nanosheet in organic solvents, and further opens a versatile platform for fabricating polymer nanocomposite materials via in situ solution polymerization.
3.2 Preparation of GO-MDI/HBPEIs nanocomposites
As designed, the obtained GO-MDI is employed to prepare GE-MDI/HBPEI nanocomposites with various GO-MDI contents by in situ random solution co-polycondensation or crosslinking reaction, and subsequent synchronous thermal imidization and reduction. Because that there are plenty of various oxygen-containing active functional groups on the surface of each GO nanosheet,18 the number of MDI moieties linked to the GO nanosheet with free isocyanato end groups is varied. But the rough content of nitrogen atom in the isocyanato group on the surface of GO-MDI nanosheet can be estimated of 4.5 wt% as mentioned above.As shown in Scheme 2(a), GO-MDI is used as a multi-functional co-monomer to prepare (GE-MDI)-co-HBPEI nanocomposites with a novel triamine (TAPPP), commercial diamine (ODA) and dianhydride (BPADA), via in situ random solution co-polycondensation and subsequent synchronous thermal imidization and reduction. Meanwhile, GO-MDI is used as a multi-functional crosslinker, which makes the precursor of HBPEI derived from TAPPP, ODA and BPADA, link with each other by in situ solution crosslinking reaction and the same subsequent process, to prepare (GE-MDI)-g-HBPEI nanocomposites with various GO-MDI contents, as shown in Scheme 2(b).
The surface reflection FTIR spectra of (GE-MDI)-co-HBPEI and (GE-MDI)-g-HBPEI nanocomposites with various GO-MDI contents are shown in Fig. 6(a) and (b) in turns. The FTIR results of this two kinds of nanocomposites with various GO-MDI contents are nearly the same as that of pure HBPEI. The matrixes are proved with complete imidization by the appearance of the characteristic peaks of imide at 1776 cm−1 (CO asymmetrical stretching), 1723 cm−1 (CO symmetrical stretching), 1376 cm−1 (C–N stretching) and 744 cm−1 (CO bending), together with the disappearance of peaks around 1670 cm−1 corresponding to CO stretching of amide. While the characteristic peaks of GO-MDI which is used as a co-monomer or crosslinker, is not detected for its very low content.
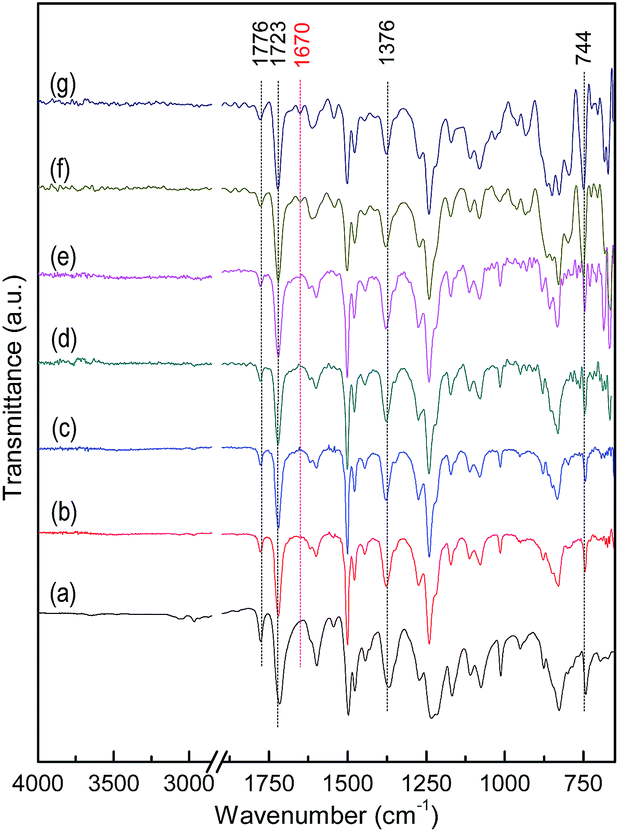 | ||
| Fig. 6 FTIR spectra of (a) pure HBPEI, (GE-MDI)-co-HBPEI nanocomposites with GO-MDI contents: (b) 0.25 wt%, (c) 0.50 wt%, (d) 1.0 wt% and (GE-MDI)-g-HBPEI nanocomposites with GO-MDI contents: (e) 0.25 wt%, (f) 0.50 wt%, (g) 1.0 wt%. | ||
As the chemical structure of the obtained nanocomposites is illustrated by FTIR spectrum, the physical aggregation morphology is illustrated according to the XRD pattern in Fig. 7. The pure HBPEI shows a broad peak at around 17.5°, revealing its amorphous nature. In addition, the incorporation of GE-MDI nanosheets into the HBPEI backbones with various GO-MDI contents does not significantly change the amorphous nature of the two kinds of GE-MDI/HBPEI nanocomposites. In addition, no peak of GO-MDI was detected at 9.3°, thus suggesting the GO-MDI nanosheets were fully exfoliated and homogeneously dispersed in the polymer matrix.38 This is mainly because that the covalent interaction which exists at the molecular level between the matrix and different contents of additive restricts the orientation of the polymer chains and modified GO nanosheets. It is also probably due to the negligible amount of GO which do not get up to the sensitivity.50,51
 | ||
| Fig. 7 XRD patterns of (a) pure HBPEI, (GE-MDI)-co-HBPEI nanocomposites with GO-MDI contents: (b) 0.25 wt%, (c) 0.50 wt%, (d) 1.0 wt% and (GE-MDI)-g-HBPEI nanocomposites with GO-MDI contents: (e) 0.25 wt%, (f) 0.50 wt%, (g) 1.0 wt%. | ||
3.3 Properties of GE-MDI/HBPEI nanocomposites
| Sample | Tg (°C) | Storage modulus at 50 °C (GPa) | T5% (°C) | T10% (°C) | Rwc (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| DSCa | DMTAb | N2 | Air | N2 | Air | ||||
a Tg values obtained by DSC in N2.b Tg values obtained by DMTA with the peak temperature in tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ curves in air.c Residual weight percentages at 800 °C in N2 as obtained by TGA.d Not measured. δ curves in air.c Residual weight percentages at 800 °C in N2 as obtained by TGA.d Not measured. |
|||||||||
| 0 wt% | 216.9 | 225.0 | 1.65 | 538.1 | 491.0 | 561.4 | 529.3 | 63.8 | |
| (GE-MDI)-co-HBPEI | 0.10 wt% | —d | — | — | 542.4 | 488.5 | 562.8 | 540.4 | 63.3 |
| 0.25 wt% | 225.8 | 232.3 | 1.72 | 540.3 | 495.7 | 566.8 | 536.6 | 63.2 | |
| 0.50 wt% | 228.7 | 237.1 | 1.94 | 541.6 | 500.7 | 569.0 | 537.6 | 62.5 | |
| 0.75 wt% | 233.3 | — | — | 544.6 | 489.1 | 571.2 | 546.3 | 62.2 | |
| 1.0 wt% | 235.5 | 243.1 | 2.11 | 546.6 | 490.2 | 572.8 | 546.9 | 61.7 | |
| (GE-MDI)-g-HBPEI | 0.10 wt% | — | — | — | 542.7 | 496.2 | 562.6 | 536.5 | 63.6 |
| 0.25 wt% | 222.8 | 225.4 | 1.78 | 541.9 | 497.9 | 563.0 | 538.9 | 63.0 | |
| 0.50 wt% | 225.4 | 228.3 | 1.84 | 544.5 | 502.2 | 566.4 | 541.4 | 62.5 | |
| 0.75 wt% | 228.3 | — | — | 545.1 | 496.5 | 564.9 | 543.4 | 62.3 | |
| 1.0 wt% | 231.4 | 231.4 | 1.98 | 543.8 | 499.5 | 568.4 | 543.3 | 61.7 | |
The heat resistance of material indicates its physical tolerance on the temperature. The glass-transition temperature (Tg), which is one of the important indicators to characterize the heat resistance, is generally considered to be the ultimate-use temperature of structural polymer material. As shown in Fig. 8, the Tg values of the resulting two kinds of GE-MDI/HBPEI nanocomposites with various GO-MDI contents, obtained by the second heating trace of the DSC curves in N2. Compared with that of the corresponding pure HBPEI, the Tg values of these two kinds of nanocomposites are all increased by a decade or two degrees, which depends on the content of the GO-MDI. For one thing, GO-MDI which is used as a multi-functional co-monomer or crosslinker, makes the network structure of the nanocomposite more compact and stable, and restricts the movement of polymer chains. For another, the modified GE nanosheet which has outstanding thermal conductivity, is introduced into the matrix polymer by covalent bond. The good compatibility and interfacial interaction between the modified GE nanosheet and the matrix polymer, is advantageous to the thermal conduction and diffusion. While the Tg values of GE/PI nanocomposites as Qian23 and Yoonessi32 reported, have little change compared with that of corresponding pure PI.
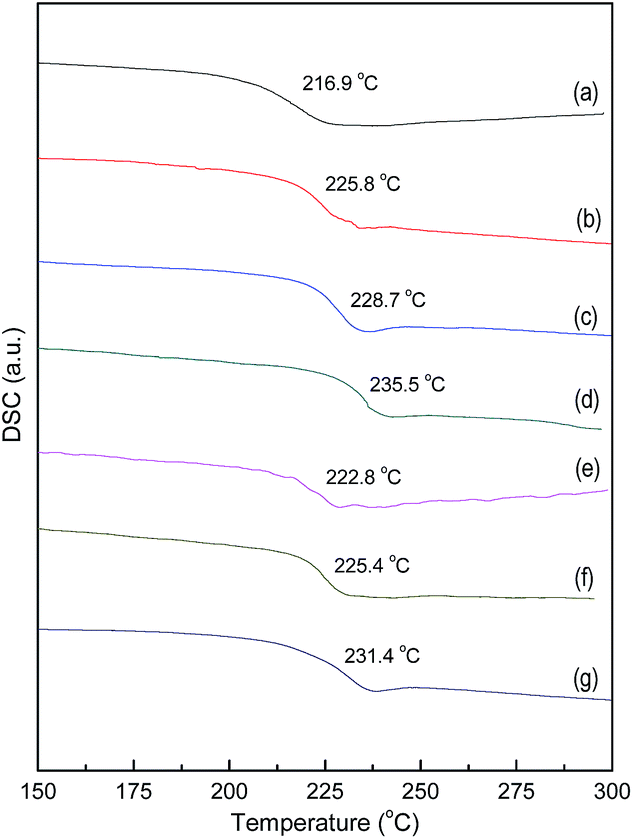 | ||
| Fig. 8 DSC curves of (a) pure HBPEI, (GE-MDI)-co-HBPEI nanocomposites with GO-MDI contents: (b) 0.25 wt%, (c) 0.50 wt%, (d) 1.0 wt% and (GE-MDI)-g-HBPEI nanocomposites with GO-MDI contents: (e) 0.25 wt%, (f) 0.50 wt%, (g) 1.0 wt%. | ||
In addition, the Tg values of (GE-MDI)-co-HBPEI nanocomposites are a little higher than that of the corresponding (GE-MDI)-g-HBPEI nanocomposites. It is mainly because that the linear imide segment between the adjacent modified GE-MDI nanosheets of the former ones is generally shorter than that of the later ones which are obtained by the crosslinking of precursors (AD-HBPAA) with a certain chain length. As a result, the flexibility of polymer chains of the formers is naturally less than that of the laters.
DMTA is generally used to evaluate the viscoelasticity of polymeric material, and another effective means of measuring the Tg value. From the DMTA measurement of the obtained two kinds of GE-MDI/HBPEI nanocomposites with various GO-MDI contents in air Fig. 9(a) and (b), the Tg values which are obtained by the peak temperature of the tanδ-temperature curves, are also gradually increased with increasing the content of GO-MDI. Furthermore, the Tg values are all slightly higher than that obtained by DCS method, due to their different measuring principles.
 | ||
| Fig. 9 DMTA measurement of the obtained (a) (GE-MDI)-co-HBPEI and (b) (GE-MDI)-g-HBPEI nanocomposites with various GO-MDI contents in air. | ||
As shown in Fig. 9(a) and (b), the storage modulus at 50 °C of pure HBPEI is 1.65 GPa, and the storage moduli of the two kinds of nanocomposites gradually increase with GO-MDI content increasing. The reason also relates to GO-MDI which is used as a multi-functional co-monomer or crosslinker in the polymerization system. It not only improves the compatibility and interfacial interaction between the host and guest, but also makes the crosslinked network structure of the nanocomposite much more compact and stable. This rigid crosslinked network structure results in that the brittleness of the nanocomposite increases continually with the increase of GO-MDI content.
TGA trace is also one of the main methods to evaluate the thermal stability of high-temperature polymer, with the indicators including temperatures for 5% weight loss (T5%) and 10% weight loss (T10%). The TGA curves of the obtained two kinds of GE-MDI/HBPEI nanocomposites with various GO-MDI contents in N2 are presented in turn in Fig. 10(a) and (b). From the figures, it is found that T5% and T10% values of pure HBPEI in N2 are 538.1 °C and 561.4 °C respectively, exhibiting good thermal stability. The thermal stability of the nanocomposites, with both T5% and T10% in N2 shows an increasing trend with the increase of GO-MDI content from 0.10 wt% to 1.0 wt%. This results are consistent with those of the GE/PI nanocomposites, which was reported by Qian23 and Zhang.38 This is partly because that the crosslinked network structure formed by covalent interaction between the polymer matrix and modified GE nanosheet is beneficial for the diffusion or transfer of heat. On the other hand, the carbon surface of the modified GE nanosheet in the polymer matrix might act as a radical scavenger to inhibit radical formation and delay the pyrolysis process.52 However, the residual weight percentage (Rw) of the obtained two kinds of nanocomposites at 800 °C in N2 is found to be a little smaller (≤2.1%) than that of pure HBPEI. This is partly associated with the thermolysis of the residual oxygen-containing species and oxygenated graphic carbon skeleton on the GO-MDI nanosheets, as well as the decomposition of amido and urethane linkages in the polymer matrix. Furthermore, the molar ratio of the functional groups which participate in the co-polycondensation, is gradually close to a equimolar state and is broke again with further addition of GO-MDI. The redundant unreacted groups are also one of the critical influencing factors. In addition, the small molecular gases in the microvoids which were formed by thermolysis of the residual oxygen-containing species on GO-MDI nanosheets during the preparation process,18,34 are released rapidly when heated to molten state.
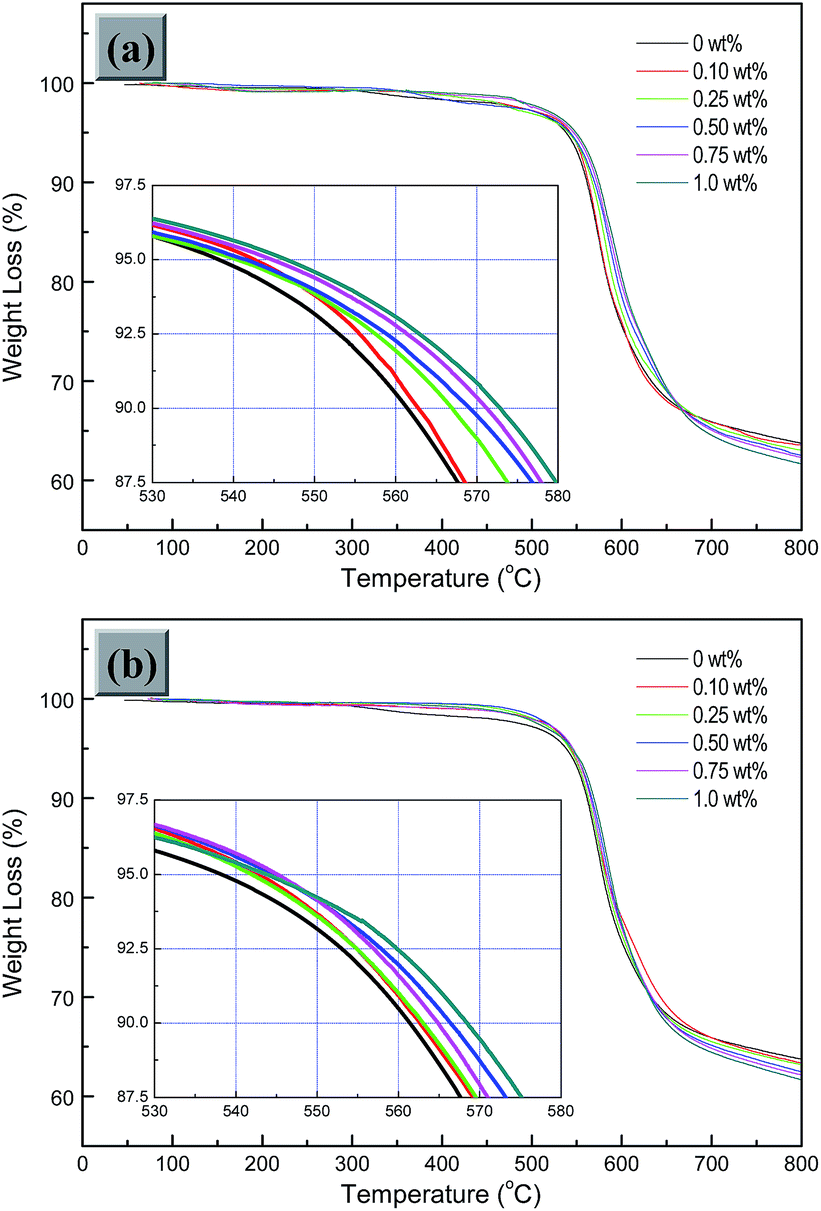 | ||
| Fig. 10 TGA curves of the obtained (a) (GE-MDI)-co-HBPEI and (b) (GE-MDI)-g-HBPEI nanocomposites with various GO-MDI contents in N2. | ||
As shown in Fig. 11(a) and (b), T5% and T10% of the obtained two kinds of nanocomposites with various GO-MDI contents in air are generally higher than that of pure HBPEI, with the biggest increase close to 20 °C. This is basically the same as the result reported by Liu.25 By contrast, the thermal stability data of the obtained two kinds of nanocomposites in air are twenty or thirty degrees lower than those in N2, due to the oxidation and other correlated reaction of the polymers in the presence of oxygen. In addition, the weight of the obtained two kinds of nanocomposites in air drops to zero definitely in the range of 690–750 °C, and pure HBPEI in air is completely weightless to zero at 675 °C, ascribing to the aerobic combustion of polymer in air. This is also the well-known reason that the thermal stability under air atmosphere is widely called as the thermo-oxidative stability.
 | ||
| Fig. 11 TGA curves of the obtained (a) (GE-MDI)-co-HBPEI and (b) (GE-MDI)-g-HBPEI nanocomposites with various GO-MDI contents in air. | ||
 | ||
| Fig. 12 Typical stress–strain curves of the obtained (a) (GE-MDI)-co-HBPEI and (b) (GE-MDI)-g-HBPEI nanocomposites with various GO-MDI contents. | ||
 | ||
| Fig. 13 Changes of the (a) tensile strength and tensile modulus, (b) elongation at break of the obtained two kinds of GE-MDI/HBPEI nanocomposites with increasing the content of GO-MDI. | ||
 | ||
| Fig. 14 SEM photographs of fracture surface of the obtained (GE-MDI)-co-HBPEI nanocomposites after tensile test with GO-MDI contents: (a): 0 wt%, (b): 0.10 wt%, (c): 0.25 wt%, (d): 1.0 wt% and (GE-MDI)-g-HBPEI nanocomposites after tensile test with GO-MDI contents: (e): 0.10 wt%, (f): 0.25 wt%, (g): 1.0 wt%. | ||
To be specific, the pure HBPEI presents a typical mechanical characteristic of engineering plastics with high strength (80.5 MPa), high modulus (1.38 GPa), low elongation at break (12.5%), as well as no yield point in the stress–strain curve. From Fig. 14(a), the SEM photograph of fracture surface of pure HBPEI after tensile test is quite smooth and flat, which fits well with the typical deformed veins of rigid material.
For (GE-MDI)-co-HBPEI nanocomposites, when the content of GO-MDI is 0.10 wt%, the average tensile strength, tensile modulus and elongation at break values of are 96.6 MPa, 1.56 GPa and 13.1%, respectively. Compared with pure HBPEI, the increasing percent of the three parameters are 20.0%, 13.0% and 4.8%, respectively. The mechanical enhancement is mainly because that GO-MDI is used as a multi-functional co-monomer to be introduced into the HBPEI backbone by covalent bonding. It not only improves the interfacial compatibility and interaction between the modified GE nanosheet and HBPEI matrix, but also makes the network structure of the nanocomposite much more compact and stable, further restricts the oriented movements of polymer chains when stretched. Furthermore, the modified GE nanosheets in this kind of nanocomposites, which are obtained by in situ random solution co-polycondensation, are randomly dispersed in the HBPEI matrix without any ordered accumulation or arrangement. As shown in Fig. 15, these disorderly dispersed nanosheets could be stretched in the direction of plane, or flipped at local scope. This is one of the primary reasons that the mechanical toughness of this nanocomposite is slightly increased when the addition amount of GO-MDI is very small. Such positive promotion is evidenced from the SEM photograph of fracture surface of (GE-MDI)-co-HBPEI nanocomposite with 0.10 wt% GO-MDI after tensile test in Fig. 14(b). It is relatively much rougher, and the deformation veins on the SEM photographs of fracture surface are relatively more denser and irregular compared with that of pure HBPEI. With the content of GO-MDI gradually increasing to 1.0 wt%, the average tensile strength and tensile modulus values of this nanocomposites dramatically increase to 130.4 MPa and 1.95 GPa, with the increasing percentage up to 62.0% and 41.3% respectively. But the average elongation at break value falls sharply to 8.5%, only 68.0% of that of the pure HBPEI. This is because that the addition of GO-MDI although increases the crosslinking density and elasticity, as well as enhances the rigidity. In addition, the modified GE nanosheets are inevitable to stack or aggregate closer when their loadings exceed a certain amount. It would reduce the surface area of the filler, and more importantly the stacked or aggregated.
 | ||
| Fig. 15 The changes of modified GE nanosheets dispersed in the nanocomposite before and after stretching. | ||
GE nanosheet is more likely to be caused with local deformation, slippage and even crack under stress. It is called mechanical percolation53 that the mechanical property of composite is obviously deteriorated when the filler addition exceeds a critical level. As shown in Fig. 14(c) and (d), varying degrees of protuberances on the SEM photographs of fracture surface of (GE-MDI)-co-HBPEI nanocomposites with 0.25 wt% and 1.0 wt% GO-MDI after tensile test, are more obvious and intensive. Moreover, some irregular cracks and interstices are also observed and they show the same change rules. One of the incentives of these irregular cracks or interstices, is the local slip or split of the aggregated GE-MDI nanosheets when stretching, and another is the number and size of these microvoids and other defects formed during the preparation process increase with increasing the content of GO-MDI, resulting in the adjacent ones connecting with each other. And of course, the changes of the molar ratio of the reactants directly affects the molecular weight of the whole polymer, which is also one of the reasons for the above mechanical properties change.
For (GE-MDI)-g-HBPEI nanocomposites, when the content of GO-MDI increases from 0 to 1.0 wt%, the average tensile strength and tensile modulus values progressively increase to 114.9 MPa and 1.78 GPa, with the increasing percent of 42.7% and 29.0% in order compared with those of pure HBPEI. While the average elongation at break value shows a changing rule of downward parabola. The maximum is up to 16.3% (the increasing percent is 30.4%) with 0.25 wt% GO-MDI, and the last is just 10.4% (83.2% of that of pure HBPEI). The mechanical properties of this nanocomposites varies depending on the content of GO-MDI, which is used as a multi-functional crosslinker. It increases the crosslinking density of the composite system, which positively impacts on the rigidity and strength. But excess addition also results in the structural defects of the nanocomposite more and more obvious with the mechanical toughness deteriorating ceaselessly. The corresponding SEM photographs of fracture surface of (GE-MDI)-g-HBPEI nanocomposites with 0.10 wt%, 0.25 wt% and 1.0 wt% GO-MDI after tensile test (Fig. 14(e)–(g)), show the similar appearances characteristics as those of (GE-MDI)-co-HBPEI nanocomposites.
By contrast, it also found that the average tensile strength and tensile modulus values of (GE-MDI)-co-HBPEI nanocomposite are slightly higher than those of the corresponding (GE-MDI)-g-HBPEI nanocomposite, while the average elongation at break value of the former is a little lower than the corresponding latter. This is because the chain length and flexibility of the polymer chain between the neighbouring modified GE nanosheets in the latter nanocomposite which is obtained via the crosslinking of the precursor polymer with a certain molecular weight, are all different degrees of greater or better than those of the former one.
It was found that the introduction of layered materials (such as clay, LDH) in PI matrix could reduce the gas permeability rate56 generally due to their high aspect ratio and good alignment in polymer matrix.57 Recently, GE or its modified layered product were also proved to be an effective candidates of gas or moisture barrier materials for various polymer matrix.58–60 In this work, taking two common micromolecular gases with different molecular weights (N2 and H2) as examples, the gas permeability results of the resulting two kinds of GE-MDI/HBPEI nanocomposite films are shown in Fig. 16. As shown in this figure, P(N2) and P(H2) of pure HBPEI film are 3.933 × 10−15 cm3 cm cm−2 s−1 Pa−1 and 345.0 × 10−15 cm3 cm cm−2 s−1 Pa−1 respectively. Significant decreases (more than 40%) in P(N2) and P(H2) of this two kinds of nanocomposite films are observed after adding 0.1 wt% of GO-MDI in the above HBPEI matrix. The two data almost fall with increasing GO-MDI content until 0.5 wt%, since then they both rise slightly but still lower than those of pure HBPEI film. The modified GE nanosheets in this two kind of nanocomposites with high aspect ratio and high specific surface area, might be more inclined to be aligned with the film surface due to the solution flow and solvent evaporation during the solution casting process,19 and further could effectively extend the path of the micromolecule gas passing through the thin film.57 However, the water vapor transmission rate (WVTR) of the nanocomposite films which were obtained by simple solution mixing of GE and PI, or their precursors,57,58 were remarkably reduced (more than 80%) compared to that of pure PI. It was because the flexible HBPEI led to a relative weaker incompact packing of barrier layers on the matrix, resulting in a relative moisture permeability than rigid PI matrix. Furthermore, excess GO-MDI content results in some adverse microvoids in the nanocomposites which is derived from the stack or aggregation of modified GE nanosheets and thermolysis of the residual oxygen-containing species,18,34 and thus affects their gas barrier properties with unusually deteriorations.
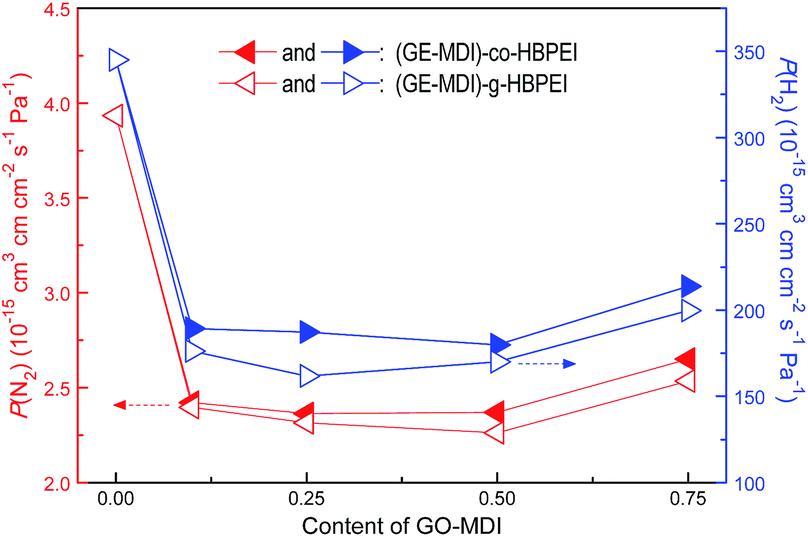 | ||
| Fig. 16 Effect of GO-MDI content on N2 and H2 permeability coefficient (P(N2) and P(H2)) of the resulting two kinds of nanocomposite films. | ||
In addition, the P(N2) and P(H2) of (GE-MDI)-co-HBPEI nanocomposite films are slightly higher than those of (GE-MDI)-g-HBPEI nanocomposite films with the same GO-MDI content. On the one hand, this is because that the modified GE nanosheets in the HBPEI matrix of the former are dispersed more random and intensive, leading to the structural defects are relatively more than the later. On the other hand, the relative length and flexibility of the latter polymer chain between the neighbouring modified GE nanosheets are longer or better than that of the former, resulting in the physical intertwining and interaction between these segments being stronger, and further making their arrangement and stacking more closely.
4 Conclusion
An effective approach to prepare two kinds of GE-MDI/HBPEI nanocomposites via in situ random solution co-polycondensation or crosslinking reaction, as well as subsequent synchronous thermal imidization and reduction, is demonstrated with highly enhanced thermal, mechanical and gas barrier properties. Previously, high concentration and completely exfoliated GO colloidal dispersion in NMP obtained by solvent-exchange method, provides convenience for subsequent preparation of high-quality GO-MDI, which is suitable for the further solution polymerization. The chemical structure and composition, the microstructure as well as the relevant thermoanalysis are characterized to confirm that GO is modified successfully for GO-MDI with free isocyanato groups. The chemical structure and physical aggregation morphology of these nanocomposites with various GO-MDI contents are also studied by some relevant means. With the increase of GO-MDI content, Tg, T5% and T10% values of the obtained two kinds of nanocomposites increase gradually by a few degrees to a decade or two degrees, and the mechanical strength and modulus of them also increase continually with the biggest increasing percentage of 42.7% and 29% or even higher, respectively, but their mechanical toughness present the characteristics of downward parabola with growths of the maximum 4.8% or 30.4% for the different types. In addition, gas barrier properties for N2 or H2 show a significant improvements with P(N2) and P(H2) values decreasing about 40% under a small GO-MDI content. Furthermore, except that mechanical toughness and gas barrier properties of the nanocomposites obtained by in situ random solution co-polycondensation are less than those of the corresponding nanocomposites obtained by in situ random solution crosslinking reaction, the other properties of the formers are all different degrees of superior to those of the laters. This work develops a facile and effective way to fabricate high performance GE/PEI nanocomposites, and lays some foundations for enriching the variety or developing multi-function or multi-purpose of PEI-based nanocomposites, which can meet the usage requirements of advanced engineering or functional materials.Acknowledgements
This work was financially supported by the Natural Science Foundation of Hubei Province (2013CFB007) and National Natural Science Foundation (51272071). The authors also acknowledge Ministry-of-Education Key Laboratory for the Green Preparation and Application of Functional Materials and Prof. Y.-B. He for providing necessary facilities.References
- M. K. Ghosh and K. L. Mittal, Polyimides: Fundamentals and Applications, Marcel Dekker, New York, 1996 Search PubMed.
- G. Wypych, PEI poly(ether imide), Handbook of Polymers, 2012, pp. 359–363 Search PubMed.
- D.-J. Liaw, K.-L. Wang, Y.-C. Huang, K.-R. Lee, J.-Y. Lai and C.-S. Ha, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef CAS.
- M. Kakimoto and M. Jikei, Prog. Polym. Sci., 2001, 26, 1233–1285 CrossRef.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS.
- K. Yamanaka, M. Jikei and M. Kakimoto, Macromolecular, 2000, 33, 6937–6944 CrossRef CAS.
- J.-J. Hao, M. Jikei and M. Kakimoto, Macromolecules, 2002, 35, 5372–5381 CrossRef CAS.
- Y. Liu and T. S. Chung, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4563–4569 CrossRef CAS.
- H. Chen and J. Yin, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3804–3814 CrossRef CAS.
- J.-J. Hao, M. Jikei and M. Kakimoto, Macromol. Symp., 2003, 199, 233–242 CrossRef CAS.
- H. Gao, D. Wang, S.-W. Guan, W. Jiang, Z.-H. Jiang, W.-N. Gao and D.-M. Zhang, Macromol. Rapid Commun., 2007, 28, 252–259 CrossRef CAS.
- T. Itoh, S. Gotoh, T. Uno and M. Kubo, J. Power Sources, 2007, 174, 1167 CrossRef CAS.
- J. Peter, A. Khalyavina, J. Kříž and M. Bleha, Eur. Polym. J., 2009, 45, 1716–1727 CrossRef CAS.
- J. Xia, G. Zhang, L. Deng, H. Yang, R. Sun and C.-P. Wong, RSC Adv., 2015, 5, 19315–19320 RSC.
- C.-M. Leu, G. M. Reddy, K.-H. Wei and C.-F. Shu, Chem. Mater., 2003, 15, 2261–2265 CrossRef CAS.
- X. Jia, Q. Zhang, M.-Q. Zhao, G.-H. Xu, J.-Q. Huang, W. Qian, Y. Lu and F. Wei, J. Mater. Chem., 2012, 22, 7050–7056 RSC.
- H. Bai, C. Li and G. Shi, Adv. Mater., 2011, 23, 1089–1115 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. Chen, H. Zhu and T. Liu, ACS Appl. Mater. Interfaces, 2010, 2, 3702–3708 CAS.
- W.-H. Liao, S.-Y. Yang, S.-T. Hsiao, Y.-S. Wang, S.-M. Li, H.-W. Tien, C.-C. M. Ma and S.-J. Zeng, RSC Adv., 2014, 4, 51117–51125 RSC.
- T. Huang, R. Lu, C. Su, H. Wang, Z. Guo, P. Liu, Z. Huang, H. Chen and T. Li, ACS Appl. Mater. Interfaces, 2012, 4, 2699–2708 CAS.
- N. D. Luong, U. Hippi, J. T. Korhonen, A. J. Soininen, J. Ruokolainen, L.-S. Johansson, J.-D. Nam, L. H. Sinh and J. Seppälä, Polymer, 2011, 52, 5237–5242 CrossRef.
- Y. Qian, Y.-F. Lan, J.-P. Xu, F.-C. Ye and S.-Z. Dai, Appl. Surf. Sci., 2014, 314, 991–999 CrossRef CAS.
- X.-L. Fang, X.-Y. Liu, Z.-K. Cui, J. Qian, J.-J. Pan, X.-X. Li and Q.-X. Zhuang, J. Mater. Chem. A, 2015, 3, 10005–10012 CAS.
- H. Liu, Y. Li, T. Wang and Q. Wang, J. Mater. Sci., 2012, 47, 1867–1874 CrossRef CAS.
- G. Y. Kim, M.-C. Choi, D. Lee and C.-S. Ha, Macromol. Mater. Eng., 2012, 297, 303–311 CrossRef CAS.
- J. Dong, C. Yin, X. Zhao, Y. Li and Q. Zhang, Polymer, 2012, 54, 6414–6424 Search PubMed.
- J.-Y. Wang, S.-Y. Yang, Y.-L. Huang, H.-W. Tien, W.-K. Chin and C.-M. Ma, J. Mater. Chem., 2011, 21, 13569–13575 RSC.
- Y. Li, X.-L. Pei, B. Shen, W.-T. Zhai, L.-H. Zhang and W.-G. Zheng, RSC Adv., 2015, 5, 24342–24351 RSC.
- I. H. Tseng, J.-C. Chang, S.-L. Huang and M.-H. Tsai, Polym. Int., 2012, 62, 827–835 CrossRef.
- L. Cao, Q.-Q. Sun, H.-X. Wang, X.-X. Zhang and H.-F. Shi, Composites, Part A, 2015, 68, 140–148 CrossRef CAS.
- M. Yoonessi, Y. Shi, D. A. Scheiman, M. Lebron-Colon, D. M. Tigelaar, R. A. Weiss and M. A. Meador, ACS Nano, 2012, 6, 7644–7655 CrossRef CAS PubMed.
- W.-H. Liao, S.-Y. Yang, S.-T. Hsiao, Y.-S. Wang, S.-M. Li, C.-C. M. Ma, H.-W. Tien and S.-J. Zeng, ACS Appl. Mater. Interfaces, 2014, 6, 15802–15812 CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- S. Park, J. An, I. Jung, R. D. Piner, S. J. An, X. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS PubMed.
- L.-B. Zhang, J.-Q. Wang, H.-G. Wang, Y. Xu, Z.-F. Wang, Z.-P. Li, Y.-J. Mi and S.-R. Yang, Composites, Part A, 2012, 43, 1537–1545 CrossRef CAS.
- N. Kumar, S. M. Liff and G. H. Mckinley, US Pat. 253219, 2005.
- S. M. Liff, N. Kumar and G. H. Mckinley, Nat. Mater., 2007, 6, 76–83 CrossRef CAS PubMed.
- Y. Wang, Z. Shi, J. Fang, H. Xu and J. Yin, Carbon, 2011, 49, 1199–1207 CrossRef CAS.
- W.-Q. Chen, Q.-T. Li, Q.-Y. Zhang, Z.-S. Xu, X.-B. Wang and C.-F. Yi, J. Appl. Polym. Sci., 2015, 132, 41544 Search PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef CAS.
- Y. W. Zhu, S. Murali, W. W. Cai, X. S. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- N. D. Luong, N. Pahimanolis, U. Hippi, J. T. Korhonen, J. Ruokolainen, L. S. Johansson, J. D. Nam and J. Seppälä, J. Mater. Chem., 2011, 21, 13991–13998 RSC.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS.
- H. Wang, J. T. Robinson, X. Li and H. Dai, J. Am. Chem. Soc., 2009, 131, 9910–9911 CrossRef CAS PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- Y. C. Cao, C. X. Xu, X. Wu, X. Wang, L. Xing and K. Scott, J. Power Sources, 2011, 196, 8377–8382 CrossRef CAS.
- S. J. Park, J. H. An, J. R. Potts, A. Velamakanni and S. Murali, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- H. W. Ha, A. Choudhury, T. Kamal, D.-H. Kim and S.-Y. Park, ACS Appl. Mater. Interfaces, 2012, 4, 4623–4630 CAS.
- X. Zhao, Q. Zhang, D. Chen and P. Lu, Macromolecules, 2010, 43, 2357–2363 CrossRef CAS.
- Y. H. Kim and O. W. Webster, J. Am. Chem. Soc., 1990, 112, 4592–4593 CrossRef CAS.
- Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685–1698 CrossRef CAS.
- H. Y. Huang, T. C. Huang, T. C. Yeh, C. Y. Tsai, C. L. Lai, M. H. Tsai, J. M. Yeh and Y. C. Chou, Polymer, 2011, 52, 2391–2400 CrossRef CAS.
- M.-H. Tsai, C.-J. Chang, H.-H. Lu, Y.-F. Liao and I.-H. Tseng, Thin Solid Films, 2013, 544, 324–330 CrossRef CAS.
- I.-H. Tseng, Y.-F. Liao, J.-C. Chiang and M.-H. Tsai, Mater. Chem. Phys., 2012, 136, 247–253 CrossRef CAS.
- O. C. Compton, S. Kim, C. Pierre, J. M. Torkelson and S. T. Nguyen, Adv. Mater., 2010, 22, 4759–4763 CrossRef CAS PubMed.
- H. Kim, Y. Miura and C. W. Macosko, Chem. Mater., 2010, 22, 3441–3450 CrossRef CAS.
Footnote |
| † These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |