
Superior relaxation of stresses and self-healing behavior of epoxy-amine coatings†
Abstract
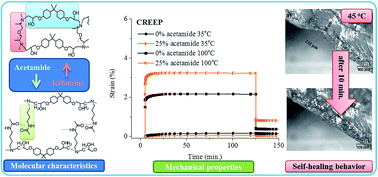
Cross-linked epoxy resins are amongst the most widely used materials for protecting metal surfaces in high performance applications. During curing or application of such materials as a coating on a metal surface, mechanical stresses can build up and ultimately lead to mechanical failure. Stress-relaxation in fully covalent systems is, however, limited due to the restricted mobility of the network chain segments. Hereby, we introduce physical cross-links (hydrogen bonds into the epoxy-amine network) via the incorporation of amide motifs, which enhance temporary local network mobility and ensure a better ability to relax the stresses preemptively, without any significant change in modulus and (dry and wet) adhesion behavior. The reversibility of hydrogen bonds also results in superior restoration of superficial scratches with no detrimental effect on the original adhesion properties of the material.
 Please wait while we load your content...
Please wait while we load your content...

