
Bifunctional catalyst of a metallophthalocyanine-carbon nitride hybrid for chemical fixation of CO2 to cyclic carbonate†
Abstract
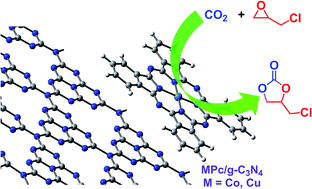
Chemical fixation of carbon dioxide (CO2) to cyclic carbonates was investigated by using bifunctional nucleophile–electrophile catalysts of a metallophthalocyanine–carbon nitride hybrid [MPc/g-C3N4 (M = Co, Cu)] in the absence of any co-catalysts and organic solvents. MPc/g-C3N4 was readily obtained by direct calcination of a mixture of dicyandiamide and metallophthalocyanine under a flowing-nitrogen atmosphere, and the MPc/g-C3N4 prepared at 480 °C (MPc/g-C3N4-480) showed the highest catalytic performance toward the cycloaddition reaction of CO2 to epichlorohydrin (ECH). For bifunctional MPc/g-C3N4, the MPc species function as Lewis acidic centers for ECH activation via electrophilic attack; while, the g-C3N4 moiety, possessing abundant and uncondensed species with the forms of primary amine (NH2) groups and secondary amine (C–NH–C) groups at the edges of graphitic sheets as edge defects, acts as an organic base for CO2 activation through nucleophilic attack. The developed MPc/g-C3N4 is stable and insoluble in any commonly used organic solvents and behaves as heterogeneous catalyst, leading to facile separation and recycling in a CO2 fixation reaction.
 Please wait while we load your content...
Please wait while we load your content...

