
Preparation of useful building blocks, α-iodo- and bromoalkanols from cyclic ethers using the Dowex H+/NaX (X = I, Br) approach†
Petri A. Turhanen* and
Jouko J. Vepsäläinen
University of Eastern Finland, School of Pharmacy, Biocenter Kuopio, P.O. Box 1627, FIN-70211, Kuopio, Finland. E-mail: petri.turhanen@uef.fi
First published on 3rd February 2016
Abstract
Our recently reported novel green chemistry tool was effectively used for opening cyclic ethers to produce α-iodo- and bromoalkanols. The synthesis of 4-iodobutanoic acid from γ-butyrolactone has also been described. The method is based on the use of a dried Dowex H+/NaX (X = Br, I)-system, which is effective at producing α-iodoalkanols and some α-bromoalkanols from commercially available cyclic ethers. Additionally, opening of three different crown ethers to form α-iodo(polyethylene)glycols with various chain lengths is demonstrated. Haloalkanols are important building blocks in synthetic chemistry e.g. for medicinal chemistry purposes.
Introduction
We have recently discovered a powerful tool with which to perform common organic addition and substitution reactions by utilizing dried solid acidic material with the simultaneous use of sodium iodide either as a catalyst or reagent.1 This report details how the same approach can be applied for the preparation of α-iodo- and bromo-alkanols from cyclic ethers.Haloalkanes and haloalkanols are important reagents and building blocks which can be used in common reactions such as alkylation of amines, etherification of alcohols and phenols, esterification of carboxylic acid salts, Michaelis–Arbuzov reactions and esterification of phosphonates.2–4 In general, iodo- and bromoalkanols are much more reactive than chloroalkanols which usually need elevated temperatures if they are to react.2,5 α-Iodoalkanols can be prepared from the corresponding chloro- or bromoderivatives by a common halogen exchange reaction (NaI/acetone).6–8 However it is a much more economical and environmentally-friendly way to prepare α-iodoalkanols directly from starting materials not containing a halogen such as cyclic ethers as will be described in here.
It is generally known that the ring opening reaction of a ring containing a saturated ether bond depends to a great extent on the ring size, because small, three or four atoms containing, rings such as oxirane and oxetane can be quite easily opened under acidic conditions. In addition, there are published examples for the preparation of 5-halopentanol from tetrahydrofuran (THF). However only a few reports can be found in the literature which describe the synthesis of 5-halopentanol from tetrahydro-2H-pyran (THP). In fact, there is only one paper which has described the synthesis of 5-iodopentanol from THP by using the toxic NaI/BF3–etherate system9 and this can be avoided by adopting our method.1 The Dowex H+/NaI approach also makes it possible to prepare 2-(2-iodo-ethoxy)-ethanol (10) from 1,4-dioxane as we have recently reported (see Scheme 1).1 Different size ethylene glycols are very important compounds with which to optimize the properties of a variety of biologically important molecules10–13 and 2-(2-iodo-ethoxy)-ethanol (10) is precursor of the diethylene glycol structure.14
 | ||
| Scheme 1 Synthesis of 2-(2-iodo-ethoxy)-ethanol (10) from 1,4-dioxane. | ||
Results and discussion
In Table 1, we have collected the tested ring opening reactions for commercially available starting materials. As shown, the isolated yields for the bromide adducts 4b and 6b are moderate but still reasonable, in fact in the case of 12b, the yield is quite good. We also tested whether bromide adducts could be made from 1, 7 and 9, but according to their 1H NMR spectra only a few per cents of corresponding products were present in the crude reaction mixtures.| Substrate | Conditionsa, yieldb | Product | Substrate | Conditionsa, yieldb | Product |
|---|---|---|---|---|---|
| a In the table, only the reaction time and temperature have been highlighted, all reactions were performed using the dried Dowex H+/NaX (X = Br, I) system, detailed experimental procedures and conditions can be found in the ESI.b Isolated yields.c Crude yield, ca. 91% purity.d Reported earlier.1e Conversion. | |||||
 |
50 °C, 18 h, 27%, (55%)c |  |
 |
50 °C, 18 h, 53% (4a), 29% (4b) |  |
 |
70 °C, 18 h, 86% (6a), 34% (6b) | 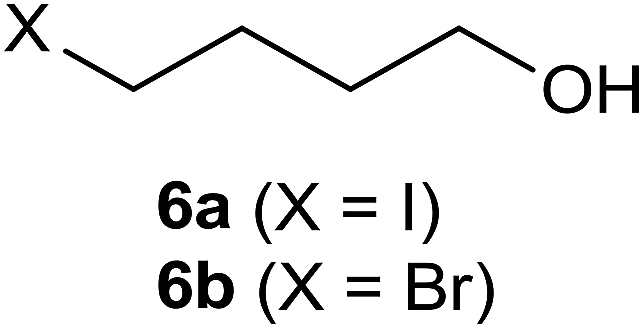 |
 |
92 °C, 18 h, 47% |  |
 |
105 °C, 18 h, 68%d |  |
 |
60 °C, 18 h, 75% (12a), 59% (12b) |  |
 |
122 °C, 18 h, 36%, (100%)e |  |
 |
142 °C, 24 h, 33%, (95%)e |  |
 |
142 °C, 24 h, 32% |  |
 |
142 °C, 24 h, 22% | 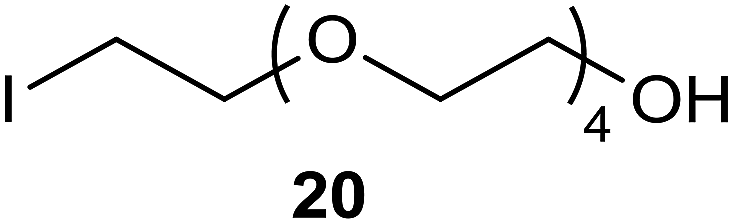 |
 |
142 °C, 24 h, 14% |  |
|||
In the case of the iodide adducts, the isolated yields were generally reasonable or even high. Compound 2 was isolated with a 27% yield (the yield of crude product was 55% and purity ca. 91%, although adequate for its further use in most cases), which was quite low, probably due to its rather low boiling point (volatility) and instability. Compounds 4a, 6a, 8, 10 and 12a were isolated at moderate to high yields, the most interesting cases were the degradation of energetically stable six-membered rings 7 and 9 which we demonstrated earlier, but now we were able to improve the isolated yield of 8 from 33% to 47%.1 Compound 12a was isolated with a high 75% yield; this is a very interesting building block along with 12b for optimizing molecular properties e.g. for medicinal chemistry use, since many drugs have a cyclohexyl structure.15,16 Interestingly, those building blocks have been sparingly reported in the literature, e.g. experimental NMR spectral data were not available and according to a SciFinder search, these compounds are not commercially available. The 100% and 95% conversions of the compounds 13 to 14 and 15 to 16, respectively, were observed according to NMR samples measured from crude reaction mixtures. However, the isolated yield was only 36% and 33%, respectively, because of the presence of a “back reaction” (see Experimental section). The degradation of 2,3-dihydrobenzofuran (13) makes the method attractive because of its potential use in the destruction of samples containing chlorinated dibenzofurans which are extremely stable and highly toxic chemical compounds.17 The significance of different size ethylene glycols and the ability of our method to synthesize 10 from 1,4-dioxane (9) led us to test if we could degrade also crown ethers (17, 19 and 21) and produce corresponding ethylene glycol precursors (18, 20 and 22). When crown ethers (17, 19 and 21) were refluxed in xylene (ca. 142 °C) for 24 h, products 18, 20 and 22 were isolated with 32%, 22% and 14% yields, respectively. Although these yields are not impressive, one has to bear in mind that in the method reported here, only one reaction step is needed, compared to the earlier method in the literature.18 Furthermore, if one has to use either pentaethylene glycol or hexaethylene glycol as starting materials for the synthesis of compounds 20 and 22 according to the method reported in the literature18 then the synthesis will be a much more costly procedure; these materials are about 3.5 and 2.5 times more expensive than starting from the corresponding crown ethers 19 and 21, respectively (in fact, compound 20 has not been reported earlier according to SciFinder). In addition, should the intent be to conduct a preliminary search for the optimal length of the ethylene glycol spacer without the need to synthesize more than 10–100 mg of the precursor, this method is definitely a convenient option. The main reason for the low yields of the products 18, 20 and 22 is believed be the degradation of the desired products during the reaction, since we were able to isolate some of degradation products from the reaction mixture (as an example see the compounds isolated in the reaction mixture of 19 in Fig. 1). All the isolated compounds 18, 23 and 24 were confirmed by examining the 1H, 13C NMR and MS spectra. It is proposed that our method is also capable of degrading straight ethers, the formation of compound 24 was not unexpected because the method can be used for the direct substitution of a hydroxyl group into iodide as we have reported earlier.1
 | ||
| Fig. 1 Some of the side-products isolated from the reaction mixture of 19. | ||
The choice of the most appropriate solvent to be used in these reactions was far from straightforward. Our earlier experiences with the reaction system used here indicated that alkylnitriles such as acetonitrile and butyronitrile would be the best solvents to be used in all of these ring opening reactions, however this was not the case. It would have been theoretically possible to investigate the effect of different solvents individually in each of the reactions reported here, but that would be beyond the scope of this article. Many of the reported reactions were tested with at least two different solvents and the solvent which achieved the best yields is described in the Experimental section in the ESI.† As an example, there were very little differences in the isolated yields between acetonitrile and acetone in the synthesis of 2. In the case of 2a, acetonitrile was better than 2-propanol, however, in the synthesis of 2b the use of 2-propanol achieved a better yield. In the syntheses of 6, 8 and 10, no additional solvents were used, because the starting materials (5, 7, and 9) are commonly used solvents and thus they were successfully exploited as such. Interestingly, when compounds 12a–b were synthesized using acetone as the solvent, higher yields were obtained than with acetonitrile (crude yields were around the same as when using acetonitrile). This was quite unexpected because acetone readily forms a self-condensation product under these kinds of acidic conditions, a fact confirmed from inspection of the 1H NMR spectrum of the crude product (self-condensation of acetone was observed also in the synthesis of 2). In the synthesis of 14, butyronitrile was used as the solvent, achieving a 100% conversion yield, whereas acetonitrile resulted in only 41% conversion. Probably the main reason for this difference is the lower temperature which can be used with acetonitrile (acetonitrile b.p. 82 °C and butyronitrile b.p. 118 °C). Higher temperatures were needed to reach ca. 95% conversion of γ-butyrolactone (15) to 4-iodobutanoic acid (16) and for this reason xylene was selected as the solvent in this reaction. All the crown ether ring opening reactions were performed also in xylene, because the use of butyronitrile resulted in lower yields. In summary, it proved difficult to identify any direct relationship between the solvent used in the reactions reported here other than a temperature correlation. More strained rings needs less energy (lower temperatures) than more stable rings (higher temperatures).
We also tested whether different equivalent amounts of NaI or NaBr should be used in the reactions to obtain the best yields; the amounts are reported individually for all reactions in the Experimental section in the ESI† as are the amounts of Dowex H+ used in these reactions.
Conclusions
Our very recently reported method1 was successfully applied to prepare highly adaptable building blocks to be used in synthetic chemistry with only one reaction step, starting from cyclic ethers (or an ester in the case of 16). This new approach is much more environmentally-friendly and economical than published methods because the Dowex H+ resin can be regenerated and reused as we have earlier reported1 and there are no need to use already halogenated starting materials (halogen exchange reaction) and no need for the use of toxic reagents such as NaI/BF3–etherate system.9 The reported yields for the prepared compounds ranged from 14% to 86%, and although these could probably be improved by optimizing the reaction conditions, this would be a time-consuming process, since it needs to be done on a case-by-case basis.Acknowledgements
This research has been supported by strategic funding from the University of Eastern Finland. The authors would like to thank Mrs Maritta Salminkoski for her expert technical assistance and Dr Jukka Leppänen and Mrs Miia Reponen for performing the MS measurements. Mrs Tiina Koivunen is acknowledged for her help with the elemental analyses.Notes and references
- P. A. Turhanen and J. J. Vepsäläinen, RSC Adv., 2015, 5, 26218 RSC.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms and Structure, John Wiley & Sons, Inc., 6th edn, 2007 Search PubMed.
- P. Savignac and B. Iorga, Modern Phosphonate Chemistry, CRC Press, 2003 Search PubMed.
- P. A. Turhanen and J. J. Vepsäläinen, Synthesis, 2001, 633 CrossRef CAS.
- N. S. Isaacs, Physical Organic Chemistry, Prentice Hall, 6th edn, 1995 Search PubMed.
- M. Arseneault, I. Levesque and J.-F. Morin, Macromolecules, 2012, 45, 3687 CrossRef CAS.
- P. R. Bohländer and H.-A. Wagenknech, Eur. J. Org. Chem., 2014, 34, 7547 CrossRef.
- M. Guisán-Ceinos, R. Soler-Yanes, D. Collado-Sanz, V. B. Phapale, E. Buñuel and D. J. Cárdenas, Chem.–Eur. J., 2013, 19, 8405 CrossRef PubMed.
- Z. Li, D. Wei-Guo and Z. Cheng-Han, Org. Lett., 2011, 13, 3538 CrossRef PubMed.
- K. Sano, T. Nakajima, K. Miyazaki, Y. Ohuchi, T. Ikegami, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2013, 24, 811 CrossRef CAS PubMed.
- R. Watanabe, K. Sato, H. Hanaoka, T. Harada, T. Nakajima, I. Kim, C. H. Paik, A. M. Wu, P. L. Choyke and H. Kobayashi, ACS Med. Chem. Lett., 2014, 5, 411 CrossRef CAS PubMed.
- S. S. Banerjee, N. Aher, R. Patil and J. Khandare, J. Drug Delivery, 2012, 103973 Search PubMed.
- D. A. Sheik, L. Brooks, K. Frantzen, S. Dewhurst and J. Yang, ACS Nano, 2015, 9, 1829 CrossRef CAS PubMed.
- P. K. Poutiainen, T. Rönkkö, A. E. Hinkkanen, J. J. Palvimo, A. Närvänen, P. Turhanen, R. Laatikainen, J. Weisell and J. T. Pulkkinen, Bioconjugate Chem., 2014, 25, 4 CrossRef CAS PubMed.
- F. W. Muregi and A. Ishih, Drug Dev. Res., 2010, 71, 20 CAS.
- I. Chopra and M. Roberts, Microbiol. Mol. Biol. Rev., 2001, 65, 232 CrossRef CAS PubMed.
- https://en.wikipedia.org/wiki/Polychlorinated_dibenzofurans and references therein, accessed June 25, 2015.
- J. M. Song, A. M. DiBattista, Y. M. Sung, J. M. Ahn, R. S. Turner, J. Yang, D. T. S. Pak, H.-K. Lee and H.-S. Hoe, Experimental Neurobiology, 2014, 252, 105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures for all prepared compounds and 1H NMR spectra for crude product and conversion of 2 and conversions of 14 and 16. 1H and 13C NMR spectra for the compounds 12a–b, 18, 20 and 22–24. See DOI: 10.1039/c5ra20813k |
| This journal is © The Royal Society of Chemistry 2016 |