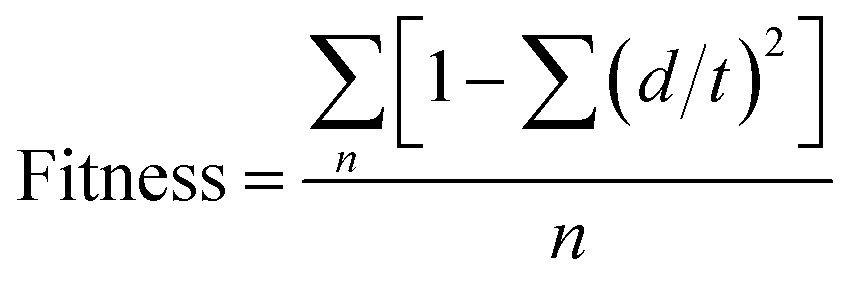DOI:
10.1039/C5RA20612J
(Paper)
RSC Adv., 2016,
6, 1466-1483
Identifying the structural features and diversifying the chemical domain of peripherally acting CB1 receptor antagonists using molecular modeling techniques†
Received
17th October 2015
, Accepted 14th December 2015
First published on 17th December 2015
Abstract
Cannabinoid 1 (CB1) receptors are a potential target for the treatment of obesity due to their role in food intake regulation and energy balance. Adverse effects associated with rimonabant, a CB1 receptor antagonist, have encouraged researchers to make efforts to design peripherally acting selective CB1 receptor antagonists. In the present work, pharmacophore mapping studies on diverse classes of compounds such as imidazoles, purines, pyrazines, piperazines, pyrazoles and pyrazolines have been performed to get structural insights into the CB1 receptor antagonists having peripheral activity. A four featured pharmacophore model has been developed having two aromatic rings, one hydrophobic center and one hydrogen bond acceptor group. A statistically robust 3D-QSAR model was developed using the pharmacophore hypothesis having a correlation coefficient of R2 = 0.990, survival score of 5.606, F value = 389, PLS factor = 4 and standard deviation of 0.164. The developed 3D-QSAR model showed excellent predictive accuracy having a correlation coefficient Q2 = 0.938. Rpred2 and Rm2 values for the test set compounds were 0.925 and 0.843 respectively. In order to diversify the limited chemical landscape of the existing peripherally acting CB1 receptor antagonists, virtual screening of the Asinex database having 435![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 214 compounds was carried out. The developed four featured pharmacophore model (AHRR), molecular docking, Lipinski's rule of five etc. were utilized as filters in the virtual screening to identify the novel hits. The most promising hits obtained through virtual screening have been reported as peripherally acting CB1 receptor antagonists. Some of these identified hits (V1, V4, V7, V8, V11, V12 and V14) have no chemical similarity to the existing class of CB1 receptor antagonists.
214 compounds was carried out. The developed four featured pharmacophore model (AHRR), molecular docking, Lipinski's rule of five etc. were utilized as filters in the virtual screening to identify the novel hits. The most promising hits obtained through virtual screening have been reported as peripherally acting CB1 receptor antagonists. Some of these identified hits (V1, V4, V7, V8, V11, V12 and V14) have no chemical similarity to the existing class of CB1 receptor antagonists.
1 Introduction
In recent years, obesity has become a primary health threat for the entire world affecting the quality of life due to the increased prevalence of a sedentary life style.1,2 Obesity, a complex metabolic disorder, involves many physiological systems such as an imbalance in energy homeostasis, abnormal development of adipose tissue, and deregulation of hormones and cytokines including adipocytokines.3 A number of chronic diseases are also associated with obesity such as type II diabetes, osteoarthritis, dyslipidemia, sleep apnea, hypertension, stroke and certain types of cancer.3,4 According to WHO, obesity is the fifth leading risk factor for global deaths.5 Thus, obesity cannot be considered a mere cosmetic issue rather it has become a major health issue.
Current approaches for the management of obesity include behavioural, dietary, pharmacological and surgical.6 Growing body of evidence has proved that obesity cannot be cured by short-term dietary control or exercise alone rather it requires additional pharmacological intervention.7 Although several drug targets have been identified till date, but only a few drugs are available for the treatment of obesity. Orlistat, a pancreatic lipase inhibitor was approved in 1999, lorcaserin, a selective 5-HT2C receptor agonist was approved in 2012, Qsymia, a combination of phentermine and topiramate was approved in July 20128 and most recently in September 2014, FDA approved contrave (bupropion and naltrexone)9 for the treatment of obesity. Orlistat is a moderately effective drug causing gastrointestinal adverse effects while lorcaserin was initially rejected in preclinical studies in 2010 by FDA due to carcinogenicity. Another drug sibutramine, a dual serotonin norepinephrine reuptake inhibitor, was withdrawn in 2010 due to increased risk of cardiovascular side effects.10 Rimonabant (a), a centrally acting cannabinoid 1 (CB1) receptor antagonist developed as an anti-obesity agent was also withdrawn from the market due to its psychiatric side effects in 2008.10–13 Thus, only a few drugs could hit the market for the treatment of obesity, that too with significant drawbacks. Hence, the market demands effective anti-obesity drugs having lesser side effects for the treatment of obesity.
Endocannabinoids bind and activate the cannabinoid (CB) receptors. Two types of cannabinoid receptors have been identified i.e. CB1 and CB2. CB1 receptors are expressed widely throughout the body, mainly located in the brain regions such as basal ganglia, cerebellum, hippocampus etc. and in the peripheral tissues such as adipose tissues, pituitary and adrenal glands, gastrointestinal tract, heart, sympathetic ganglia, lungs, urinary bladder and liver.4,14 Over the past one decade, the roles of cannabis and endocannabinoids in the regulation of appetite have been studied extensively.15 Endocannabinoid system contributes mainly to the physiological regulation of food intake, lipid and glucose metabolism and energy balance through central as well as peripheral effects.14
As discussed earlier, rimonabant (a) was withdrawn due to serious psychiatric side effects such as anxiety, depression and even suicidal tendency. A new strategy, considered to be more effective and safer for counteracting obesity, is the development of peripherally restricted CB1 receptor antagonists, which would be devoid of psychiatric side effects. URB447 (b), the first peripherally restricted mixed CB1 antagonists/CB2 agonist having an efficacy comparable to rimonabant for reducing body weight gain and food intake was synthesized in 200916 but during experimentation, the drug URB447 (b) was found to penetrate in the CNS. Compound AM6545 (c) was reported as a non-brain-penetrant neutral CB1 receptor antagonists having lesser lipid solubility with high affinity and selectivity towards CB1 receptors as compared to rimonabant. Compound AM6545 showed brain/plasma concentration ratio of 0.03 which indicated markedly reduced brain penetration of the drug in CNS.17 A novel series of alkynylthiophene derivatives were designed by 7TM Pharma company and their efforts led to identification of TM38837 (d) as a first-in-class second generation peripherally acting CB1 receptor antagonist having very good potency (EC50 = 8.5 nM) and less CNS penetration with low brain-to-plasma ratio (B/P = 1/33).18 Recently, 7TM Pharma company has reported that in Phase 1 clinical trial study, compound TM38837 was restricted to the periphery of the human body.19,20 Thus, these reports substantiate the observations that the designing of peripheral acting selective CB1 receptor antagonists could be an important approach for the treatment of obesity.21,22
Review of literature4,21 on the available CB1 receptor antagonists also revealed the fact that the existing set of CB1 receptor antagonists have a limited chemical diversity as all these reported compounds have chemically similar scaffolds. There is a need to discover newer CB1 receptor antagonists possessing entirely new scaffolds so that the domain of discovering newer CB1 receptor antagonists could expand.
Ligand-based drug designing approaches such as pharmacophore and QSAR23,24 are based on the assumption that all the compounds interacting with a common target would share similar structural or physicochemical properties. Structural features such as aromatic ring, hydrophobic group, H-bond donor or H-bond acceptor groups etc. are important components of a pharmacophore model. These features are useful for characterization of structurally diverse compounds interacting with the same target.25 In the discovery of novel lead compounds, pharmacophore modelling and 3D-QSAR techniques are powerful tools which may shed light on the essential structural features required for eliciting the desired biological activity. Various computational techniques such as 3D-QSAR, pharmacophore mapping, homology modeling, docking and virtual screening have been utilized for designing of CB1 receptor antagonists in the last few years. All these studies have been directed for centrally acting CB1 receptor antagonists.26–44 Due to the adverse effects reported with centrally acting compounds, designing of selective peripherally acting CB1 receptor antagonists is considered as a novel task. Thus, some molecular modeling techniques were applied for designing of peripherally acting CB1 receptor antagonists.22 In our previous publication,22 we have reported the 3D-QSAR (CoMFA/CoMSIA) studies on 1,5-diaryl pyrazole containing CB1 receptor antagonists only having peripheral activity. In the present work, a wide array of structures have been used having involving six different scaffolds i.e. imidazoles, purines, pyrazines, piperazines, pyrazoles and pyrazolines to find out the common essential structural features for the peripherally acting CB1 receptor antagonists. The present study utilizes techniques like pharmacophore modeling, 3D-QSAR modeling, molecular docking and virtual screening. The information so derived has been utilised for the identification of the promising hits as peripherally acting CB1 receptor antagonists to broaden the chemical landscape of the currently existing narrow chemical spectrum of peripherally restricted CB1 receptor antagonists so that newer CB1 receptor antagonists can be discovered having lesser side effects and better therapeutic efficacy for the control of obesity. The present study is a step towards the designing of newer unknown chemical classes of peripherally acting CB1 receptor antagonists. Here, both the ligand and the structure based drug design approaches are combined together to mine more reliable information which could be successfully applied to develop novel peripherally acting CB1 receptor antagonists.
Identification of pharmacophore from a diverse set of compounds is considered to be an important step in rational drug designing approaches. A number of diverse classes of peripherally acting CB1 receptor antagonists have been reported that showed lesser degree of side effects. It was planned to gain insight into the structural requirements responsible for peripherally restricted CB1 receptor antagonists.
One important aim of the current study was to infuse and introduce newer chemical diversity to CB1 receptor antagonists in terms of entirely different scaffolds than the existing ones, it was also decided to undertake virtual screening of a sufficiently large chemical database. The developed pharmacophore and 3D-QSAR models were used further for the identification of novel hits through a virtual screening protocol. Only the peripherally acting compounds were used for the development of the pharmacophore model. A diverse set of compounds bearing imidazole, purine, pyrazine, piperazine, pyrazole and pyrazolines as the basic scaffolds have been used for the development of the pharmacophore model. Compounds are picked up on the basis of structural diversity and variations in biological activity. The alignment obtained from the pharmacophoric features was used to further develop an atom-based 3D-QSAR model to identify and understand the essential chemical features required for peripherally acting CB1 receptor antagonists. A four point pharmacophore model was identified having two aromatic rings, one hydrophobic center and one hydrogen bond acceptor group as essential structural features. An excellent predictive 3D-QSAR model was also derived by aligning all peripherally acting CB1 receptor antagonists of the set according to the identified pharmacophore features. The generated pharmacophore features, molecular docking, Lipinski's rule of five, developed 3D-QSAR, CNS score and receptor ligand interactions were used as filters to perform virtual screening of Asinex database containing 435214 compounds for the identification of novel peripherally acting CB1 receptor antagonists. Finally, the most promising 14 hits so obtained have been reported as peripherally acting CB1 receptor antagonists.
2 Experimental sections
2.1 Selection of dataset
Various classes of selective CB1 receptor antagonists have been reported in the last few years. Initially, a set of 190 compounds from six different classes such as imidazole, purine, pyrazine, piperazine, pyrazole and pyrazolines were chosen from the literature.45–56 Thus, the first criterion for the selection of the compounds was selectivity for peripheral receptors. Selectivity of CB1 receptor over CB2 receptor was also considered. Compounds having human CB1 receptor activity only were considered in the present study. Compounds whose stereochemistry and biological activity were not clearly defined were omitted from the study. Further, selections of the compounds were made on the basis of deviation in the biological activity and structural variations. The most active, moderately active and least active compounds of each scaffold were considered so that a robust data set could be prepared. For avoiding redundancy of information, compounds having similar biological activity and chemical structures were omitted from the final data set. 13 compounds were removed due to ambiguous stereochemistry and biological activities. Finally, a set of 35 compounds were selected for the present study. Rest of the compounds (S1–S142) were used as an additional external set for further validation purpose. Biological activities of all these compounds were reported as Ki values (nanomolar) which were converted into their corresponding pKi values by taking negative logarithm of Ki values (pKi = −logKi). The dataset so obtained was divided into a model-building set containing 25 compounds (1–25) as shown in Fig. 1 and an external test set of 10 compounds (26–35) which were used for the validation of the developed model as shown in Fig. 2. The pKi values of all the selected compounds for developing the pharmacophore model were ranging from 5.074 to 10.046. For the 3D-QSAR modeling, the model-building set was further divided into a training set and test set in the ratio of 4:1. The threshold pKi values for actives and inactives were fixed to 7.5 and 6.6 respectively to get 8 active, 8 inactive and 9 moderately active compounds which were utilised for the development of pharmacophore model and the subsequent scoring function.
 |
| | Fig. 1 Chemical structures of compounds (1–25) used for the development of pharmacophore model. | |
 |
| | Fig. 2 External test set compounds (26–35) used for validation. | |
Pharmacophore hypothesis was developed using PHASE QSAR module of Schrodinger that employs a grid-based 3D-QSAR analysis.57 Grid points nearest to the atoms in the molecule aligned to a selected reference compound were identified. Thus, the grid points so obtained were used to generate 3D descriptors for different types of interactions to correlate with the biological activities using PLS analysis.58,59 Chemical structures of all the compounds were sketched and cleaned using ‘building tools’ option in Maestro molecular modeling software. All the sketched molecules were optimized using the LigPrep version 2.3.60 The optimized molecules were imported into PHASE for the development of common pharmacophore hypothesis (CPH) and the associated alignment. Conformers of all the compounds were generated using ConfGen search method with OPLS-2005 force field. A maximum of 1000 conformers per structure with 100 conformers per rotatable bond were generated by a preprocess minimization of 100 steps and a distance-dependent dielectric solvation treatment was applied.
2.2 Development of pharmacophore model
A set of pharmacophore features for the selected compounds (1–25) were produced using create sites option. Site points were created for each conformer of the compounds under study. For creating pharmacophore sites, a default setting having aromatic ring (R), hydrophobic (H), positive (P), H-bond donor (D), and acceptor (A) features were used. Common pharmacophore hypothesis for the set of active ligands were generated using these features. A set of variants formed by a set of features was used to identify the common pharmacophore using a tree based partitioning algorithm with a criterion that the selected variants must match with all the active compounds. The generated common pharmacophore of all variants were scored for the active and inactive compounds to identify a set of hypothesis having the best alignment of all the actives. The common pharmacophore hypotheses so obtained were scored by setting the root mean square deviation (RMSD) value below 1.0 and vector score value to 0.5.61 “Score hypothesis” was the last step in the pharmacophore development where hypotheses were ranked to make rational choices among the generated hypotheses. The most appropriate hypothesis was selected for further exploration. All the generated pharmacophore hypothesis were ranked accordingly on the basis of their statistical parameters such as survival score and ‘survival minus inactive’ (S − I).62,63 Survival score for the active compounds is the weighted combination of the volume, site, vector, survival score and a term for the number of matches. The survival score and S − I correspond to ‘score active’ and ‘score inactive’ respectively. In addition to this, post hoc score was also used to validate the best pharmacophore hypothesis. The highest value of ‘score active’ of the pharmacophore model has the ability to identify active molecules whereas the highest value of ‘score inactive’ of the model has the ability to segregate the active and inactive molecules.63 Survival score (S) is the final scoring function which can be represented with the following eqn (1):| | |
S = WsiteSsite + WvecSvec + WvolSvol + WselSsel + Wrewm − WEΔE + WactA
| (1) |
where, S represents the scores and W represent weights. Ssite, Svec, Svol and Ssel denote alignment score, vector score, volume score and selectivity score respectively. Ssite is the RMSD in the site point position, Svec is the cosine of the angles formed by corresponding pairs of vector features in aligned structures and Svol is based on overlap of van der Waals models of nonhydrogen atoms in each pair of structures. Default values of Wsite, Wvec, Wvol were 1.0 whereas default value of Wsel was 0.0. Wrewm represents the reward in which Wrewm is user-adjustable (1.0 by default) where m denotes the number of actives that match the hypothesis minus one. WEΔE denotes penalty for high-energy structures by subtracting a multiple of the relative energy from the final score and it penalizes a hypothesis for which the reference ligand activity is lower than the highest activity by adding a multiple of the reference ligand activity to the score denoted by WactA, where A denotes the activity.64 Higher survival score indicates better mapping of the pharmacophore with the active ligands and the fitness score confirms the quality of the pharmacophore hypothesis that can be defined as how well the compounds could be mapped to a pharmacophore model which can be calculated by using the following eqn (2):| |
 | (2) |
where, n is the number of pharmacophore features, d is the displacement of the feature from the centre of the location constraint and t represent the radius of the location constraint sphere for the feature.65,66 Based on 8 active ligands in the dataset, several 3-point, 4-point and 5-point pharmacophore hypotheses were generated. Each generated common pharmacophore hypothesis resulted into alignment of all the compounds. These alignments were further used to develop 3D-QSAR models.
2.3 Development of 3D-QSAR model
In the present study, atom-based 3D-QSAR models were developed because atom-based 3D-QSAR considers the entire molecular space while pharmacophore-based 3D-QSAR does not consider the chemical space beyond the pharmacophoric groups. Thus, atom-based 3D-QSAR is more advantageous than pharmacophore-based 3D-QSAR for the development of a good QSAR model.15 The molecular alignment obtained through pharmacophore generation was used to generate 3D-QSAR model by using partial least square (PLS) factors. Maximum of n/5 PLS factors can be used where n is the number of compounds in the training set. So, in the present work a maximum 4 PLS factors were allowed. Each atom in the atom-based model served as a sphere. The training set compounds were covered with a regular grid of cubes. Each cube represented upto six “bits”, meaning six different classes of atoms including H-bond donor (D), hydrophobic or non polar (H), negative ionic (N), positive ionic (P), electron-withdrawing (W) and others (X). Several 3D-QSAR models were generated and each generated model was validated by the internal test set compounds which were randomly selected from model-building set in the ratio of 4:1 in training and test set respectively. The developed models were evaluated by different parameters such as cross-validated coefficient, noncross-validated coefficient, F-value, predictive r2, modified r2, Pearson-R etc.
For the development of QSAR model, PLS was used to linearly correlate independent variables as descriptors and dependent variable as biological activity. The internal predictive accuracy of the model was evaluated by the cross-validated coefficient using leave-one-out (LOO) method calculated by using eqn (3):
| |
 | (3) |
where,
Ypredicted,
Yactual and
Ymean are the predicted, actual and mean values of activity.
67,68
The predictive correlation coefficient (Rpredicted2) was calculated for the test set compounds by using the following eqn (4):
| | |
Rpredicted2 = SD − PRESS/SD
| (4) |
where, SD is the sum of the squared deviations between the biological activities of the test set compounds and the mean activity of the training set compounds and PRESS is the sum of squared deviation between actual and predicted activities of the test set compounds.
69
Modified r2 (rm2), an essential parameter used to validate the external predictive accuracy of the developed model, was used to penalise the model containing large difference between actual activity and the predicted activity for the test set compounds. The value of rm2 is calculated by using eqn (5):70,71
| |
 | (5) |
The developed 3D-QSAR model was accepted only if it satisfied all the following conditions: rcv2 > 0.5, rncv2 > 0.6, rpred2 > 0.5, rm2 > 0.5, [(R2 − R02)/R2] < 0.1 or [(R2 − R′02)/R2] < 0.1 and 0.85 ≤ k ≤ 1.15 or 0.85 ≤ k′ ≤ 1.15. The statistical parameters used for the validation purpose such as sensitivity, specificity, accuracy, positive prediction value (PPV), negative prediction value (NPV), Matthew's correlation coefficient (MCC) were calculated by using following eqn (6)–(11).72
| | |
Sensitivity = TP/(TP + FN) × 100%
| (6) |
| | |
Specificity = TN/(TN + FP) × 100%
| (7) |
| | |
Accuracy = (TP + TN)/(TP + FP + TN + FN) × 100%
| (8) |
| |
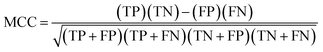 | (11) |
where, TP, FN, TN and FP are true positives, false negatives, true negatives and false positives respectively.
2.4 Virtual screening
Virtual screening of Asinex database containing 435214 diverse chemical compounds were performed with the aim of finding potential hits suitable for further development as selective CB1 receptor antagonists. Asinex database of known 3D-structures were downloaded from the official site73 and optimized using OPLS 2005 force field in LigPrep, a standard option in Maestro 9.0.60 The developed pharmacophore model was used as a first filter to search 3D database to retrieve structures that fit the pharmacophore hypothesis. Only those compounds were selected at this stage which were able to fit in all the features of the pharmacophore model. The retrieved compounds from this stage were screened further by using a second filter i.e. molecular docking studies using Glide 5.5.74 Previously reported homology model of CB1 receptor was used for the docking studies.75 Well known compounds such as rimonabant, AM6545 and TM38837 were planted in the database as reference compounds. Compounds retrieved from the first stage of filtration were docked in the active site of CB1 receptors by using high-throughput virtual screening (HTVS) method and flexible docking mode in Glide with a limit of five retained poses per molecule. Top 25000 poses were saved with fixing of van der Waals radii of ligands to 0.8 and partial charge cutoff 0.15.76 Compounds so obtained were re-docked using standard precision (SP) method for obtaining higher precision. The process was repeated again by re-docking the compounds in the active site of CB1 receptors by using extra precision (XP) method. The resulting compounds were further screened by using another filter i.e. Lipinski's rule of five. The activity of the compounds was predicted by the developed 3D-QSAR model and those compounds having activity equal to or more than 7.5 only were considered for further processing. The compounds so obtained after surviving through these filters were assessed for other properties such as polar surface area (PSA), percentage absorption, CNS score and receptor–ligand interactions. Finally, those compounds which survived in all the filtration processes were reported in the present work. The flowchart of the virtual screening is shown in Fig. 3.
 |
| | Fig. 3 Flow chart of virtual screening. | |
3 Results and discussion
3.1 Pharmacophore modeling
The present work was aimed to identify essential structural features for peripherally acting CB1 receptor antagonists. Active and inactive threshold pKi values were fixed to 7.5 and 6.6 respectively and applied to the dataset resulting into 8 active, 8 inactive and 9 moderately active compounds. For developing a common pharmacophore hypothesis, all the 8 active compounds were utilised. Pharmacophore sites were created for all the compounds to get six different kinds of structural features such as H-bond acceptor, H-bond donor, hydrophobic, positive ionizable, negative ionisable and aromatic ring. The study resulted into three common structural features i.e. H-bond acceptors, hydrophobic groups and aromatic rings with frequencies of 1, 3 and 2 respectively. The software was restricted to find a minimum of 3 and maximum of 5 pharmacophoric sites wherein a condition of 8 out of 8 active compounds must match all the sites was applied. Several 3-point, 4-point and 5-point pharmacophore hypotheses were generated on the basis of the active compounds present in the dataset. In case of 5-point hypothesis, all the active compounds could not match with all the sites. AHHRR was the most probable 5-point hypothesis. AHH, AHR, ARR, HHH, HHR, HRR and AHHH, AHHR, AHRR, HHHR and HHRR were found to be the most probable hypothesis for 3-point and 4-point pharmacophore hypothesis in which all the active compounds matched all the sites. All the generated hypotheses were evaluated. The highest survival score was used to characterize the generated hypothesis. The survival scores ranged from 2.860 to 5.606. Top ranked survival score for 3-point hypothesis was obtained for AHR.72 i.e. 3.060 whereas 4-point hypothesis AHHR.35 and AHRR.6 scored 2.860 and 5.606 respectively. Amongst all, the best hypothesis was obtained from the 4-point model (AHRR) having the highest survival score of 5.606 as shown in Table 1. The fitness scores of all the active compounds were observed for the best pharmacophore model AHRR.6. The active compound (13) was superimposed on the best model and it was observed that all the pharmacophoric features perfectly matched with a fitness score of 3.00 whereas the inactive compound (19) had a fitness score of 1.52 indicating that the developed pharmacophore model AHRR.6 has a capability to pick up the active compounds from the dataset. The best model (AHRR.6) containing one hydrogen bond acceptor, one hydrophobic group, and two aromatic rings is shown in Fig. 4. The A5 acceptor feature correctly mapped on the oxygen of carbonyl amide group attached to 3rd position of the pyrazole ring, H9 hydrophobic features mapped on the 4-Cl group of the phenyl ring at 5th position of the pyrazole ring and the two R12 and R13 aromatic rings perfectly mapped on the phenyl rings attached to 1st and 5th positions of the pyrazole ring. The distances and angles of AHRR.6 among the pharmacophore features are given in Table 2 and 3. The three top ranked pharmacophore models AHR, AHHR and AHRR were then used to align the molecules for the development of atom-based 3D-QSAR analysis. The best QSAR model was characterized by various statistical measures and regression contours. The accuracy of the developed models was verified by the statistical data such as R2 (correlation coefficient) and Q2 (cross-validated r2).
Table 1 Data of the top ranked pharmacophore models
| ID |
Survival |
Survival–inactive |
Post hoc score |
Site |
Vector |
Volume |
Selectivity |
Match |
Energy |
| AHR.72 |
3.060 |
1.260 |
3.061 |
0.63 |
0.912 |
0.519 |
1.072 |
8 |
0.710 |
| AHHR.35 |
2.860 |
1.179 |
2.864 |
0.53 |
0.893 |
0.441 |
1.411 |
8 |
1.996 |
| AHRR.6 |
5.606 |
3.527 |
3.192 |
0.72 |
0.900 |
0.572 |
1.463 |
8 |
0.164 |
 |
| | Fig. 4 Distances and angels of the best developed pharmacophore model AHRR.6. Orange coloured torus represents aromatic rings (R12 and R13), green coloured sphere represents hydrophobic group (H9), and light red coloured vector represents hydrogen bond acceptor (A5). Purple colour represents distances and green colour represents the angles between the pharmacophoric features. | |
Table 2 Distances between the different pharmacophore features of AHRR.6 model
| Site 1 |
Site 2 |
Distance (Å) |
| A5 |
H9 |
9.810 |
| A5 |
R13 |
6.846 |
| A5 |
R12 |
7.243 |
| H9 |
R13 |
3.215 |
| H9 |
R12 |
7.251 |
| R13 |
R12 |
4.719 |
Table 3 Angles between the different pharmacophoric features of AHRR.6 model
| Site 1 |
Site 2 |
Site 3 |
Angle |
| H9 |
A5 |
R13 |
8.7 |
| H9 |
A5 |
R12 |
47.4 |
| R13 |
A5 |
R12 |
39.0 |
| A5 |
H9 |
R13 |
18.8 |
| A5 |
H9 |
R12 |
47.4 |
| R13 |
H9 |
R12 |
29.3 |
| A5 |
R13 |
H9 |
152.5 |
| A5 |
R13 |
R12 |
75.1 |
| H9 |
R13 |
R12 |
131.2 |
| A5 |
R12 |
H9 |
85.2 |
| A5 |
R12 |
R13 |
65.9 |
| H9 |
R12 |
R13 |
19.5 |
3.2 Analysis of 3D-QSAR model
The alignment generated from the pharmacophore model was utilised for the development of 3D-QSAR models. Dataset was randomly divided into the training (20 compounds) and test (5 compounds) sets in a ratio of 4:1 considering uniform variation of structural diversity and biological activity of the compounds. Although a number of models were generated but only the top ranked models are discussed here. 3D-QSAR model obtained by using the 3-point pharmacophore hypothesis (AHR.72) for the purpose of alignment showed R2 = 0.967, Q2 = 0.518, SD = 0.331, PLS factor = 4, F-value = 87.2, RMSE = 0.490, pearson-R = 0.923 and stability = −0.507, while the 3D-QSAR model obtained by using the alignment on 4-point pharmacophore hypothesis (AHHR.35) showed R2 = 0.997, Q2 = 0.702, SD = 0.063, PLS factor = 4, F-value = 2435.9, RMSE = 0.523, Pearson-R = 0.852 and stability = −0.137 (Table 4). The best 3D-QSAR model was developed using the alignment based on 4-point pharmacophore hypothesis (AHRR.6) having R2 = 0.990, Q2 = 0.938, SD = 0.164, PLS factor = 4, F-value = 389, RMSE = 0.144, Pearson-R = 0.982 and stability = 0.016. Statistical parameters of the 3D-QSAR models are shown in Table 4. Predicted pKi values and residuals of the experimental (actual) and predicted pKis are shown in Table 5.
Table 4 Statistical parameters obtained for the 3D-QSAR models obtained from various alignments
| Statistical parameters |
AHR.72 |
AHHR.35 |
AHRR.6 |
| R2 |
0.967 |
0.997 |
0.990 |
| Q2 |
0.518 |
0.702 |
0.938 |
| PLS factor |
4 |
4 |
4 |
| F-Value |
87.2 |
2435.9 |
389 |
| Stability |
−0.507 |
−0.137 |
0.016 |
| RMSE |
0.490 |
0.523 |
0.144 |
| SD |
0.331 |
0.063 |
0.164 |
| Pearson-R |
0.923 |
0.852 |
0.982 |
Table 5 Experimental and predicted pKi values and the residuals of training and test set compounds by using the best developed 3D-QSAR model (AHRR.6) with fitness score
| Compd ID |
No. of conformers generated |
Experimental pKi |
Predicted pKi |
Residual |
Fitness score |
| Test set compounds. |
| 1 |
8 |
10.04 |
9.95 |
0.09 |
1.82 |
| 2a |
6 |
7.33 |
7.44 |
−0.11 |
2.07 |
| 3 |
1 |
7.52 |
7.61 |
−0.09 |
2.15 |
| 4a |
2 |
6.63 |
6.62 |
0.01 |
2.18 |
| 5 |
4 |
6.08 |
6.15 |
−0.07 |
2.14 |
| 6 |
36 |
7.85 |
7.67 |
0.18 |
1.79 |
| 7 |
9 |
5.07 |
5.38 |
−0.31 |
1.78 |
| 8 |
4 |
6.75 |
6.47 |
0.28 |
1.78 |
| 9 |
9 |
10.00 |
10.10 |
−0.10 |
2.59 |
| 10a |
14 |
7.46 |
7.63 |
−0.17 |
2.59 |
| 11 |
10 |
7.29 |
7.28 |
0.01 |
2.53 |
| 12a |
6 |
8.05 |
7.98 |
0.07 |
2.54 |
| 13 |
4 |
8.69 |
8.67 |
0.02 |
3.00 |
| 14 |
4 |
6.88 |
6.84 |
0.04 |
2.20 |
| 15 |
5 |
9.82 |
9.88 |
−0.06 |
2.08 |
| 16 |
3 |
7.25 |
7.07 |
0.18 |
1.97 |
| 17 |
3 |
6.13 |
6.42 |
−0.29 |
1.93 |
| 18 |
2 |
7.15 |
7.25 |
−0.10 |
1.57 |
| 19 |
2 |
6.14 |
6.02 |
0.12 |
1.52 |
| 20 |
13 |
5.82 |
5.84 |
−0.02 |
1.90 |
| 21 |
35 |
6.56 |
6.56 |
0.00 |
2.55 |
| 22 |
10 |
6.71 |
6.71 |
0.00 |
2.39 |
| 23a |
10 |
6.47 |
6.72 |
−0.25 |
2.47 |
| 24 |
13 |
8.92 |
9.00 |
−0.08 |
2.39 |
| 25 |
14 |
5.47 |
5.34 |
0.13 |
2.44 |
3.3 Validation of the best developed 3D-QSAR model (AHRR.6)
3.3.1 Statistical parameters. A statistically validated 3D-QSAR model is a pre-requisite for reliable predictions. Several statistical parameters such as R2, Q2, SD, RMSE, F-value etc. can be used for the evaluation of a reliable and robust model. Higher R2 and Q2 value of 0.990 and 0.938 respectively revealed that the developed 3D-QSAR model possessed high internal predictive capability. [(R2 − R02)/R2] and [(R2 − R′02)/R2] values obtained were 0.016 and 0.007 respectively which were less than 0.1. The values of k and k′ were obtained as 1.011 and 0.988 respectively (Fig. 5a and b). The F-value for the best 3D-QSAR model was 389 [F05 (4,19) = 4.500 (Tab)] at 99% confidence level which indicated that the developed model was statistically highly significant. The SD and RMSE values of 0.164 and 0.144 respectively were quite near to each other. High Pearson-R value (0.982) indicated a good ranked correlation between the experimental and predicted activity for the test set compounds. Predictive R2 (Rpred2) for the test set compounds was found to be 0.925 and Rm2 = 0.843 indicated robustness of the model. The best developed 3D-QSAR model was further validated by predicting the activity of external set of ten compounds. The experimental pKi, predicted pKi and residuals of the external test set compounds (26–35) indicated that the predicted activities are very close to the actual activities (Table 6). On the basis of threshold value (active > 7.5, inactive < 7.5), sensitivity of the best model was observed as 88.89% indicating that 88.89% of compounds were correctly predicted “actives” out of the total “actives” whereas specificity was observed as 100% which indicated that 100% of compounds were correctly predicted “non-actives” out of the total “inactives”. Accuracy of the best developed model was observed as 96% which referred to the proportion of correctly predicted as “true active” or “true inactive”. PPV was observed as 100% indicting the probability of correct positive prediction whereas NPV was observed as 94.12% indicating the probability of correct negative prediction. MCC was observed as 91.46% which measured the quality of the binary classification, indicating very good prediction. Thus, it is revealed from the statistical parameters that a reliable model has been successfully developed.
 |
| | Fig. 5 (a) Graph of experimental versus predicted activity of test set compounds, (b) graph of predicted versus experimental activity of test set compounds. | |
Table 6 External test set of compounds (26–35) used for validation
| Compd |
Experimental pKi |
Predicted pKi |
Residual |
| 26 |
7.924 |
8.125 |
−0.201 |
| 27 |
7.538 |
7.571 |
−0.033 |
| 28 |
8.215 |
7.798 |
0.417 |
| 29 |
5.415 |
6.128 |
−0.713 |
| 30 |
9.268 |
9.068 |
0.200 |
| 31 |
9.000 |
8.996 |
0.004 |
| 32 |
8.119 |
8.127 |
−0.008 |
| 33 |
7.013 |
7.330 |
−0.317 |
| 34 |
8.022 |
8.140 |
−0.118 |
| 35 |
8.301 |
8.498 |
−0.197 |
3.3.2 Additional validation of the model using external data set. External data set containing 142 compounds (S1–S142), which were not included in the model development process, were used for the true validation of the best developed model. The predictive capability of the best developed model was further evaluated by comparing the predicted activities with the experimental ones. The predicted activities of the external set compounds (S1–S142) were quite close to the experimental activities. The experimental and predicted activities of the compounds (S1–S142) are shown in Table S1 (ESI†). The graph plotted between the experimental and predicted activities of the external set compounds is shown in Fig. 6. The correlation coefficient R2 was observed as 0.687 indicating that the developed model has good predictive accuracy for predicting the biological activity of the non-evaluated compounds.
 |
| | Fig. 6 Graph of experimental versus predicted activities of external set compounds (S1–S142). | |
3.3.3 Applicability domain (AD). It is not possible always to predict the correct activity of all the compounds for any QSAR model even if it is validated with several parameters.22 AD is defined as “the response and the chemical structure space in which the QSAR model makes predictions with a given reliability”.77 It is used to determine the compounds that are outside the AD and identifying the outliers from the training set compounds. AD was calculated for training, test, external test set compounds and the virtual screening hits by using the programme “AD using standardization approach”.77 The obtained results indicated that there were no outliers present in the training set and it was also observed that the test set, external test set and hits obtained from virtual screening were inside the AD affirming the reliability of the developed 3D-QSAR model.
3.4 Graphical interpretation of the developed 3D-QSAR model
The contour maps obtained from the best 3D-QSAR model can provide insights into the structural features required for CB1 receptor antagonistic activity. The effects associated with the positive and negative regression coefficients of H-bond donor, hydrophobic, negative ionic, positive ionic, electron-withdrawing and other features could be visualized by PHASE. Visualization of the 3D-QSAR model can be done by using “regression coefficient visualization” or “effects” from atom-based or pharmacophore-based types in the grid-based 3D-QSAR methods. In the present study, effects of each pharmacophoric feature were observed with their positive and negative regression coefficients around the pharmacophore hypothesis to identify the favourable and unfavourable regions for the activity. The positive and negative coefficients are depicted in blue-coloured and red-coloured cubes respectively in all the maps (Fig. 7–10).
 |
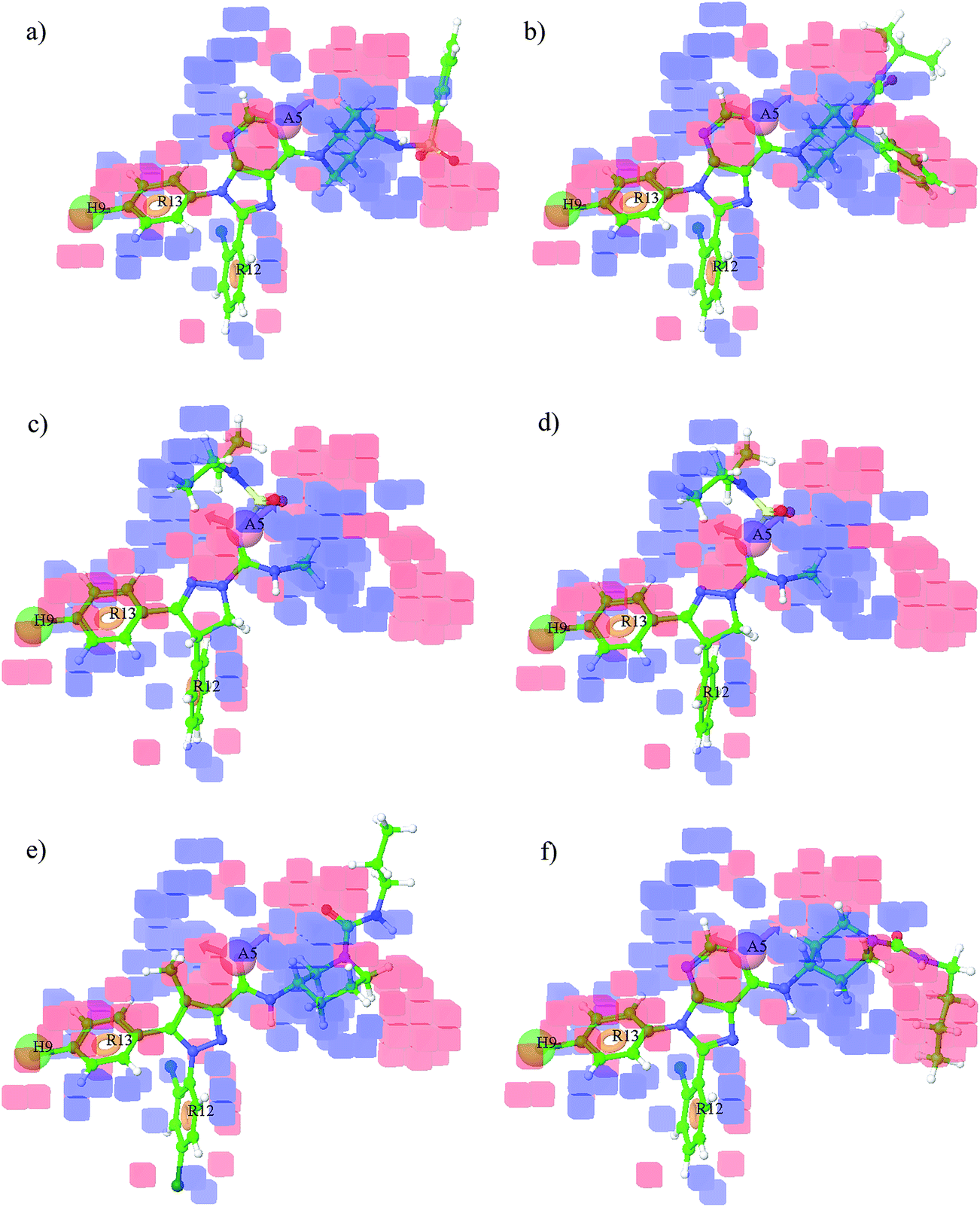
| | Fig. 7 Effects of steric substituents on (a) active compound (1), (b) inactive compound (7), (c) active compound (9), (d) inactive compound (21), (e) active compound (15) and (f) inactive compound (17). | |
 |
| | Fig. 8 Effect of electron withdrawing groups on (a) active compound (1), (b) inactive compound (7), (c) active compound (24), (d) inactive compound (23), (e) active compound (13) and (f) inactive compound (21). | |
 |
| | Fig. 9 Maps of H-bond donor effects on (a) active compound (1), (b) inactive compound (2), (c) active compound (6), (d) inactive compound (7), (e) active compound (24) and (f) inactive compound (25). | |
 |
| | Fig. 10 Effect of hydrophobic groups on (a) active compound (24), (b) inactive compound (23), (c) active compound (3), (d) inactive compound (21), (e) active compound (9) and (f) inactive compound (25). | |
3.4.1 Effect of substitution of steric (bulky) groups. With the inputs obtained from the generated contour maps due to the bulk of the groups, it was observed that steric substitution at the blue coloured cubes favoured the activity whereas substitution at the red coloured cubes disfavoured the activity (Fig. 7). The steric effects are visualized at positive and negative regression coefficient threshold of 0.006 and −0.006 respectively. Steric substituents such as tetrahydropyran ring attached to amide in the most potent compound (1) in the dataset is oriented towards blue region which is favourable for the high activity (Fig. 7a) whereas the side chain attached to the pyrazine ring in compound (7) oriented towards red region might be a reason of its poor activity (Fig. 7b). Substituent like dimethyl at 3rd position of the imidazole ring in compound (9) orientated towards the blue coloured cubes present towards right hand side of the Fig. 7c favourable region for bulkier substituents may be responsible for good activity of compound (9) whereas in compound (21), the bulkier substituent like propane attached to the 3rd position of the piperidine ring is oriented towards the red coloured cubes present at top of the Fig. 7d; this red coloured region is not favourable for the bulky substituents so this could be one of the reasons for poor activity of compound (21). Bulky substituent like cyclohexane attached to carboxamide at 4th position of the piperazine ring in compound (15) was observed near to the blue coloured cubes present towards right hand side of Fig. 7e, which favoured the bulkier substituents thus, this could be the reason of compound (15) having good activity. Pyrrolidine ring attached to the carboxamide at 4th position of the piperazine ring in compound (17) is oriented towards the red coloured cubes present at the top of the Fig. 7f. This red coloured area is unfavourable for the bulkier substituents, hence leading to poor activity for compound (17).
3.4.2 Effect of electron withdrawing groups. For understanding the effect of electron-withdrawing groups, few examples of active and inactive compounds with substituents projecting into the cubes are depicted in Fig. 8. Electron-withdrawing effects are visualized at positive and negative regression coefficient thresholds of 0.01 and −0.01 respectively. Electron withdrawing groups such as oxygen atom of carbonyl group and tetrahydropyran ring in compound (1) were present in the blue regions which are favourable for good activity (Fig. 8a) whereas oxygen of hydroxyl group of compound (7) is embedded in the red region thus offering poor activity (Fig. 8b). Oxygen atoms of the SO2 group at 4th position of the piperidine ring in compound (24) acting as electron withdrawing groups are embedded in the blue-coloured cubes (favourable region) present towards the right hand side of the Fig. 8c justifying the high activity of compound (24) whereas oxygen atom of the carbonyl group attached at 4th position of piperidine ring forming a side chain in compound (23) acts as electron-withdrawing group, and this carbonyl group embedded in the red-coloured cubes (unfavourable region) present towards right hand side of the Fig. 8d might be responsible for poor activity of compound (23). Electron-withdrawing group like cyano at 4th position of pyrazole ring in compound (13) oriented towards blue coloured cubes present at upper left hand side of the Fig. 8e as well as nitrogen of the pyrrolidine ring attached at 3rd position of the pyrazole ring side chain embedded in the blue coloured cubes present at the centre of the Fig. 8f could be responsible for high activity of compound (13). On the other hand, electron-withdrawing group like oxygen of carbonyl group attached to 3rd position of the piperidine ring in compound (21) is embedded in the red coloured cubes present towards left hand side of the Fig. 8f, which disfavoured the electron-withdrawing group in this area could be one of the reasons for compound (21) exhibiting poor activity.
3.4.3 Analysis of H-bond donor effects. For the convenience and clear viewing, the H-bond donor effects are visualized at a positive coefficient threshold of 0.01 and negative coefficient threshold of −0.01. The active and inactive compounds with their H-bond donor groups are considered to explain the background behind the differences in their activity on the basis of the generated contour maps. Blue coloured cubes indicate that H-bond donors are favoured for high activity of a compound while red coloured cubes indicate unfavourable regions for the activity. Hydrogen bond donor group present in compound (1) like NH of amide in the side chain is oriented near the blue-coloured cubes present at almost centre of the Fig. 9a, a favourable region for the activity imparting high activity to compound (1) whereas NH group at 8 position of oxepine ring in compound (2) is partially embedded in the red-coloured cubes present at the centre of the Fig. 9b, an unfavourable region for activity, thus compound (2) could be expected to exhibit poor activity. In compound (6), the hydroxyl group present in the side chain attached to the 3-position of pyrazine ring acting as hydrogen bond donor is embedded in the blue coloured cubes present on the left side of the contour maps as shown in Fig. 9c could be one of the reasons for high activity of compound (6). Whereas, the hydroxyl group present in the side chain attached to 3rd position of pyrazine ring in compound (7), embedded in the red coloured cubes present at top of the Fig. 9d, an unfavourable region for H-bond donor, justifies the poor activity of compound (7). Likewise, the NH group attached to 4th position of piperidine ring in compound (24) acting as H-bond donor is covered in the blue coloured cubes at right side of the Fig. 9e, a favourable region for the activity and this could be responsible for its high activity. Whereas in compound (25), two hydrogen bond donor groups are present, in which first NH attached to the butyl side chain is embedded in the red-coloured cubes present at right hand side of the Fig. 9f and the second NH attached to 6-position of the purine ring is embedded in the red-coloured cubes present at almost centre of Fig. 9f, both unfavourable regions for the activity, hence compound (25) should have shown poor activity.
3.4.4 Effect of hydrophobic groups. Hydrophobic effects are visualized at a positive regression coefficient threshold of 0.015 and negative regression coefficient threshold of −0.015 for interpretation of the active and inactive compounds (Fig. 10). Phenyl ring attached to SO2 group in compound (24) acting as a hydrophobic group is oriented towards blue-coloured cubes present at the right side top corner of Fig. 10a having positive coefficient for the activity and this could be the reason for good activity of compound (24) whereas in compound (23), the phenyl ring attached to 4th position of the piperidine ring is oriented towards red-coloured cubes present at the right hand side of Fig. 10b leading to poor activity of the compound (23). Hydrophobic group like diethyl attached to SO2 in compound (3) is embedded in the blue-coloured cubes present at top of the Fig. 10c, a favourable region for the activity thus exhibiting good activity for compound (3), whereas the hydrophobic n-propyl group attached to amide group in compound (21) is oriented towards red coloured cubes present at right top of the Fig. 10d indicating the negative coefficient and thus, compound (21) exhibited poor activity. Hydrophobic group like phenyl attached to 1st position of imidazole ring in compound (9) embedded in the blue-coloured cubes towards right hand side of the Fig. 10e is a favourable region for the activity and this could be the reason for good activity of compound (9) whereas the butyl group attached to amide in compound (25) is oriented towards red-coloured cubes present towards right side corner of the Fig. 10f, an unfavourable region for the activity leading to poor activity of compound (25).
3.5 Virtual screening
Virtual screening is a useful technique for the identification of hits using databases available in public/private domain. Different filters were applied (Fig. 3) to get the initial hits as CB1 receptor antagonists to the Asinex database containing 435214 compounds.
3.5.1 First filter: pharmacophore features. The best validated pharmacophore hypothesis AHRR.6 generated in this study was used as the first filter to search Asinex database. A condition that all the four features (2 aromatic rings, 1 hydrophobic group, 1 hydrogen bond acceptor) of the pharmacophore hypothesis must match to the compounds was applied. A total of 139686 compounds matched the pharmacophore features present in AHRR.6. Fitness score (fixed at ≥ 1.5) is an important parameter which measures how well the ligand fits the pharmacophore. On the basis of fitness score, 49789 compounds were obtained at this stage (Fig. 3).
3.5.2 Second filter: molecular docking. Molecular docking was applied as the second filter. As, the crystal structure of CB1 receptor still remains unknown, a homology modeled structure75 was used for the docking studies. Some reference compounds such as rimonabant, AM6545 and TM38837 were also grafted in the dataset. In the docking studies, a high-throughput virtual screening (HTVS) method was used first to dock the compounds. The reference compound rimonabant showed a G-score (−7.605) which was fixed as the threshold value for the screening. A total of 3155 compounds showed G-score greater than the threshold value (−7.605) and all these compounds were selected as hits. These 3155 hits were docked again in the active site of CB1 receptor by standard precision (SP) method using the same reference compounds. At this stage, the reference compound AM6545 showed a G-score (−9.121). Out of the 3155 compounds, 262 compounds showed higher G-scores than the scores of the reference compounds. These 262 compounds were further re-docked in the active site of CB1 receptor by extra-precision (XP) method. The reference compound AM6545 showed a G-score (−10.047) which was fixed as the new threshold value. Only 86 compounds showed higher G-score than the threshold value (−10.047) in this phase of filtering process.
3.5.3 Third filter: Lipinski's rule of five. To evaluate the hits for drug like properties the third filter of Lipinski's rule of five was applied. Only 46 compounds followed Lipinski's rule of five in toto. The molecular weight of the selected leads ranged from 377.51 to 494.52, lipophilicity in between 1.87 to 4.99, H-bond donor groups ranged from 0 to 2 and H-bond acceptors were from 4 to 9 indicating that all the 46 selected compounds followed the rules of five.
3.5.4 Fourth filter: application of 3D-QSAR model to predict the activity. CB1 receptor antagonistic activity of the 46 hits obtained from the previous stage was predicted by using the developed 3D-QSAR model. A predicted activity value higher than 7.5 was considered as the threshold value for the active compounds (similar to that maintained in the development of pharmacophore model). At this stage, a total of 19 hits having predicted activity higher than the threshold value (7.5) were obtained.
3.5.5 Fifth filter: CNS score and receptor interactions. CNS score ranging from +2 to −2 indicates the presence or absence of a drug in the brain.78 CNS score of +2 indicates the presence of the drug in the brain whereas CNS score of −2 indicates its absence. This scoring was applied for the identification of peripherally active compounds which was an essential requirement for the designing of peripherally restricted CB1 receptor antagonists. CNS score was calculated for the 19 hits obtained from the previous stage of filtration. Only 14 compounds showed CNS score of −1 or −2. These 14 hits were selected finally in which all the four pharmacophore features were present and the compounds also fulfilled other essential requirements to prove to be potent and selective peripherally acting CB1 receptor antagonists. Chemical structures of the obtained 14 hits are shown in Fig. 11.
 |
| | Fig. 11 Chemical structures and predicted activity (calculated by using the developed 3D-QSAR model) of 14 hits obtained through virtual screening. | |
It was observed from the rimonabant–CB1 receptor interaction study that the carbonyl group of rimonabant formed H-bond with Lys192, 4-chlorophenyl ring was oriented towards Val196, Phe200, Trp356 and Leu360, 2,4-dichlorophenyl ring was oriented towards Trp279 and Met363, and piperidyl group interacted with Phe177, Phe189 and Trp255 residues. The identified 14 hits are having diverse scaffolds such as pyrazole, pyridine, pyrimidine, indole, phenothiazine, pyrazolo[1,5-a]pyrimidine etc. It was observed that all of the obtained hits showed good binding interactions in the active site of CB1 receptor. H-bonds were formed by these hits with Asp266, Ile267, Phen268, Ser383 and Lys192 as shown in Fig. 12. H-bond formation with Lys192 is essential for the CB1 receptor antagonistic activity as observed in rimonabant (Fig. 12a). Peripherally acting compound AM6545 also formed H-bond with Lys192 as shown in Fig. 12b. The pyrimidine ring of compound V1 is oriented towards Val196, Phe200 and Trp356 residues, phenyl ring of the indole moiety is oriented towards Trp279 and Met363 whereas the pyridone ring is oriented towards Phe177, His178, Asp266, Ile267, Lys376 and Phe379 residues. In case of compound V2, the oxazole ring is oriented towards Trp356 and Phe200, pyrazole ring is oriented towards Phe278 and Trp279 residues whereas the pyridone ring showed interactions with Phe177, Asp266, Phe279, Ala380 and His178. Thus, it is revealed from this study that the obtained hits are suitably oriented in the active site of CB1 receptors. The orientations of compounds (V1 and V2) are shown in Fig. 12c and d respectively.
 |
| | Fig. 12 Docking interactions of (a) rimonabant (b) AM6545 (c) hit (V1) (d) hit (V2) with CB1 receptor. | |
All the hits fulfilled the physicochemical requirements for a drug like compound and were lying within the acceptable range of all the desirable parameters (Table 7). One important strategy for the designing of peripheral acting CB1 receptor antagonists is to increase the polar surface area and decrease the lipophilicity so that the designed compounds do not enter into the CNS. Polar surface area of these hits was ranging from 76.71 to 111.71, which was much higher than rimonabant (PSA = 53.32). Lipophilicity of these hits was ranging from 2.24 to 4.62, much lower than rimonabant (logP = 6.11) indicating that the obtained hits were having favourable hydrophobicity for showing peripheral CB1 receptor antagonist activity.
Table 7 Calculated physicochemical properties of the obtained 14 hitsa
| Compd |
Mol. wt |
No. of HBD |
No. of HBA |
logP |
CNS score |
PSA |
No. of rotatable bonds |
HB |
% human absorption |
| HBD: hydrogen bond donor, HBA: hydrogen bond acceptor, HB: no. of hydrogen bonds formed with the receptor. |
| V1 |
431.49 |
1 |
7 |
3.43 |
−2 |
101.35 |
6 |
4 |
94.03 |
| V2 |
435.52 |
2 |
6 |
4.62 |
−2 |
111.71 |
3 |
4 |
100.00 |
| V3 |
470.54 |
1 |
7 |
4.36 |
−2 |
80.93 |
9 |
2 |
100.00 |
| V4 |
395.43 |
2 |
5 |
4.04 |
−2 |
103.60 |
5 |
3 |
100.00 |
| V5 |
404.44 |
1 |
7 |
3.85 |
−1 |
76.92 |
5 |
3 |
100.00 |
| V6 |
429.52 |
1 |
6 |
2.88 |
−2 |
97.01 |
9 |
4 |
87.69 |
| V7 |
438.45 |
1 |
7 |
3.79 |
−1 |
93.68 |
9 |
2 |
100.00 |
| V8 |
427.45 |
1 |
7 |
3.69 |
−1 |
96.06 |
6 |
3 |
94.58 |
| V9 |
415.53 |
1 |
5 |
4.17 |
−1 |
76.71 |
6 |
4 |
100.00 |
| V10 |
390.48 |
0 |
7 |
2.96 |
−1 |
80.09 |
4 |
3 |
92.12 |
| V11 |
459.46 |
2 |
7 |
3.77 |
−2 |
100.37 |
5 |
1 |
89.93 |
| V12 |
398.46 |
1 |
5 |
3.99 |
−1 |
82.83 |
6 |
1 |
100.00 |
| V13 |
401.44 |
1 |
6 |
3.77 |
−1 |
77.83 |
5 |
3 |
96.34 |
| V14 |
423.51 |
0 |
8 |
2.24 |
−1 |
83.83 |
5 |
2 |
90.66 |
| Rimonabant |
463.79 |
1 |
5 |
6.11 |
+2 |
53.32 |
2 |
1 |
100.00 |
Virtual screening offered highly diverse set of hit molecules. Some of these scaffolds (V1, V4, V7, V8, V11, V12 and V14) have never been reported to be present in CB1 receptor antagonists. This fulfilled one of the envisaged aim of performing virtual screening i.e. diversifying the existing chemical landscape of peripherally restricted CB1 receptor antagonists.
4 Conclusions
In the present study, a ligand-based pharmacophore model with four pharmacophoric features has been developed followed by the development of a 3D-QSAR model to identify the essential features required for the peripherally acting selective CB1 receptor antagonists. The best developed pharmacophore model (AHRR.6) consisted of one H-bond acceptor, one hydrophobic center and two aromatic rings. By aligning the training and test set compounds on the best pharmacophore model (AHRR.6), a predictive 3D-QSAR model was developed. The predictive power of the developed 3D-QSAR model was validated by using an external test set compounds. Biological activity of the external set of test compounds was correctly predicted by the developed 3D-QSAR model indicating that the developed model is a reliable one. The developed pharmacophore model was further used for screening of Asinex database to search for diverse chemical entities possessing peripherally acting CB1 receptor antagonist activity. Different filters such as pharmacophore features, molecular docking, Lipinski's rule of five, minimum predicted potency using 3D-QSAR, ligand–receptor interactions and CNS scoring were applied to the dataset and finally the most favourable 14 hits having potential peripherally restricted CB1 receptor antagonistic activity were found out. The obtained hits showed good interactions with the CB1 receptor and formed H-bonds with important residues such as Lys192, Asp266, Ile267, Phen278 and Ser383. The obtained hits were also evaluated for their absence or restricted presence in CNS. Virtual screening of the Asinex database has offered very interesting leads and has been able to achieve the targeted objective of discovering entirely new scaffolds which were never known to be present in the existing CB1 receptor antagonists prior to this report, especially structures V1, V4, V7, V8, V11, V12 and V14.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
Mayank Kumar Sharma is thankful to University Grants Commission (UGC), New Delhi for awarding Senior Research Fellowships (SRF) under the RFSMS-BSR programme [No. F. 7-129/2007 (BSR)]. MRY is thankful to AICTE for the award of Research Project (F. No. -8-193/RIFD/RPS/POLICY-1/2014-15). We are also thankful to Schrodinger for providing licence of Maestro 9.4.
References
- V. D. Marzo and J. P. Despres, Nat. Rev. Endocrinol., 2009, 5, 633–638 CrossRef PubMed.
- Z. Zhang and M. Wang, Acta Pharmacol. Sin., 2012, 33, 145–147 CrossRef CAS PubMed.
- M. Alvarado, J. Decara, M. J. Luque, L. Hernandez-Folgado, M. Gomez-Canas, M. Gomez-Ruiz, J. Fernandez-Ruiz, J. Elguero, N. Jagerovic, A. Serrano, P. Goya and F. R. de Fonseca, Bioorg. Med. Chem., 2013, 21, 1708–1716 CrossRef CAS PubMed.
- M. K. Sharma, P. R. Murumkar, A. M. Kanhed, R. Giridhar and M. R. Yadav, Eur. J. Med. Chem., 2014, 79, 298–339 CrossRef CAS PubMed.
- http://www.who.int/mediacentre/factsheets/fs311/en/index.html, accessed 14 July 2014.
- M. J. Bishop, J. Med. Chem., 2006, 49, 3999–4000 CrossRef CAS PubMed.
- J. Antel, P. C. Gregory and U. Nordheim, J. Med. Chem., 2006, 49, 4008–4016 CrossRef CAS PubMed.
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm312468.html, assessed 17 Dec 2014.
- http://www.drugs.com/newdrugs/fda-approves-contrave-bupropion-naltrexone-weight-management-4081.html, assessed 17 Dec 2014.
- R. J. Rodgers, M. H. Tschop and J. P. H. Wilding, Dis. Models & Mech., 2012, 5, 621–626 CAS.
- P. Erkekoglu, B. Giray and G. Sahin, FABAD J. Pharm. Sci., 2008, 33, 95–108 Search PubMed.
- C. E. Leite, C. A. Mocelin, G. O. Petersen, M. B. Leal and F. V. Thiesen, Pharmacol. Rep., 2009, 61, 217–224 CrossRef CAS PubMed.
- E. Kirilly, X. Gonda and G. Bagdy, Acta Physiol., 2012, 205, 1–20 CrossRef.
- L. V. Gaal, A. M. Rissanen, A. J. Scheen, O. Ziegler and S. Rossner, Lancet, 2005, 365, 1389–1397 CrossRef.
- B. L. Foll, J. M. Trigo, K. A. Sharkey and Y. L. Strat, Med. Hypothesis, 2013, 80, 564–567 CrossRef PubMed.
- J. LoVerme, A. Duranti, A. Tontini, G. Spadoni, M. Mor, S. Rivara, N. Stella, C. Xu, G. Tarzia and D. Piomelli, Bioorg. Med. Chem. Lett., 2009, 19, 639–643 CrossRef CAS PubMed.
- J. Tam, V. K. Vemuri, J. Liu, S. Batkai, B. Mukhopadhyay, G. Godlewski, D. Osei-Hyiaman, S. Ohnuma, S. V. Ambudkar, J. Pickel, A. Makriyannis and G. Kunos, J. Clin. Invest., 2010, 120, 2953–2966 CAS.
- M. S. Hung, C. P. Chang, T. C. Li, T. K. Yeh, J. S. Song, Y. Lin, C. H. Wu, P. C. Kuo, P. K. Amancha, Y. C. Wong, W. C. Hsiao, Y. S. Chao and K. S. Shia, ChemMedChem, 2010, 5, 1439–1443 CrossRef CAS PubMed.
- 7TM Pharma, 7TM Pharma successfully conducts clinical Phase I trial of its first in class peripheral CB1 receptor antagonist TM38837 demonstrating restriction from the human CNS, <http://7tm.com/News.aspx?M=News%26PID=5%26NewsID=58>, accessed 15 July 2014.
- S. J. Ward and R. B. Raffa, Obesity, 2011, 19, 1325–1334 CrossRef CAS PubMed.
- M. K. Sharma, P. R. Murumkar, M. A. Barmade, R. Giridhar and M. R. Yadav, Expert Opin. Ther. Pat., 2015, 25, 1093–1116 CrossRef CAS PubMed.
- M. K. Sharma, P. R. Murumkar, R. Giridhar and M. R. Yadav, Mol. Diversity, 2015, 19, 871–893 CrossRef CAS PubMed.
- K. Roy, S. Kar and R. N. Das, Understanding the Basics of QSAR for Application in Pharmaceutical Sciences and Risk Assessment, Academic Press, 2015 Search PubMed.
- K. Roy, S. Kar and R. N. Das, A Primer on QSAR/QSPR Modeling: Fundamental Concepts (Springer Briefs in Molecular Science), Springer, 2015, DOI:10.1007/978-3-319-17281-1.
- Y. Pan, Y. Wang and S. H. Bryant, J. Chem. Inf. Model., 2013, 53, 938–947 CrossRef CAS PubMed.
- D. Shire, B. Calandra, M. Delpech, X. Dumont, M. Kaghad, G. F. Le, D. Caput and P. Ferrara, J. Biol. Chem., 1996, 271, 6941–6946 CrossRef CAS PubMed.
- J. Y. Shim, W. J. Welsh, E. Cartier, J. L. Edwards and A. C. Howlett, J. Med. Chem., 2002, 45, 1447–1459 CrossRef CAS PubMed.
- M. E. Y. Francisco, H. H. Seltzman, A. F. Gilliam, R. A. Mitchell, S. L. Rider, R. G. Pertwee, L. A. Stevenson and B. F. Thomas, J. Med. Chem., 2002, 45, 2708–2719 CrossRef CAS PubMed.
- J. Z. Chen, X. W. Han, Q. Liu, A. Makriyannis, J. Wang and X. Q. Xie, J. Med. Chem., 2006, 49, 625–636 CrossRef CAS PubMed.
- E. Cichero, G. Menozzi, A. Spallarossa, L. Mosti and P. Fossa, J. Mol. Model., 2008, 14, 1131–1145 CrossRef CAS PubMed.
- H. Wang, R. A. Duffy, G. C. Boykow, S. Chackalamannil and V. S. Madison, J. Med. Chem., 2008, 51, 2439–2446 CrossRef CAS PubMed.
- N. Foloppe, N. H. Allen, C. H. Bentley, T. D. Brooks, G. Kennett, A. R. Knight, S. Leonardi, A. Misra, N. J. T. Monck and D. M. Sellwood, Bioorg. Med. Chem. Lett., 2008, 18, 1199–1206 CrossRef CAS PubMed.
- N. S. Kang, G. N. Lee and S. E. Yoo, Bioorg. Med. Chem. Lett., 2009, 19, 2990–2996 CrossRef CAS PubMed.
- M. Ye and M. I. Dawson, Bioorg. Med. Chem. Lett., 2009, 19, 3310–3315 CrossRef CAS PubMed.
- K. C. Weber, E. F. de Lima, P. H. de Mello, A. B. F. da Silva and K. M. Honorio, Chem. Biol. Drug Des., 2010, 76, 320–329 CAS.
- N. Foloppe, K. Benwell, T. D. Brooks, G. Kennett, A. R. Knight, A. Misra and N. J. T. Monck, Bioorg. Med. Chem. Lett., 2009, 19, 4183–4190 CrossRef CAS PubMed.
- G. N. Lee, K. R. Kim, S. H. Ahn, M. A. Bae and N. S. Kang, Bioorg. Med. Chem. Lett., 2010, 20, 5130–5132 CrossRef CAS PubMed.
- S. Han, F. F. Zhang, X. Xie and J. Z. Chen, Eur. J. Med. Chem., 2014, 74, 73–84 CrossRef CAS PubMed.
- D. Hurst, U. Umejiego, D. Lynch, H. Seltzman, S. Hyatt, M. Roche, S. McAllister, D. Fleischer, A. Kapur, M. Abood, S. P. Shi, J. Jones, D. Lewis and P. Reggio, J. Med. Chem., 2006, 49, 5969–5987 CrossRef CAS PubMed.
- O. M. H. Salo, M. Lahtela-Kakkonen, J. Gynther, T. Jarvinen and A. Poso, J. Med. Chem., 2004, 47, 3048–3057 CrossRef CAS PubMed.
- C. Montero, N. E. Campillo, P. Goya and J. A. Paez, Eur. J. Med. Chem., 2005, 40, 75–83 CrossRef CAS PubMed.
- S. Durdagi, M. G. Papadopoulos, P. G. Zoumpoulakis, C. Koukoulitsa and T. Mavromoustakos, Mol. Diversity, 2010, 14, 257–276 CrossRef CAS PubMed.
- D. Latek, M. Kolinski, U. Ghoshdastider, A. Debinski, R. Bombolewski, A. Plazinska, K. Jozwiak and S. Filipek, J. Mol. Model., 2011, 17, 2353–2366 CrossRef CAS PubMed.
- R. Ai and C. A. Chang, J. Mol. Graphics Modell., 2012, 38, 155–164 CrossRef CAS PubMed.
- J. H. M. Lange, M. A. W. van der Neut, A. J. M. Borst, M. Yildirim, H. H. van Stuivenberg, B. J. van Vliet and C. G. Kruse, Bioorg. Med. Chem. Lett., 2010, 20, 2770–2775 CrossRef CAS PubMed.
- A. Fulp, K. Bortoff, Y. Zhang, H. Seltzman, J. Mathews, R. Snyder, T. Fennell and R. Maitra, J. Med. Chem., 2012, 55, 10022–10032 CrossRef CAS PubMed.
- B. A. Ellsworth, Y. Wang, Y. Zhu, A. Pendri, S. W. Gerritz, C. Sun, K. E. Carlson, L. Kang, R. A. Baska, Y. Yang, Q. Huang, N. T. Burford, M. J. Cullen, S. Johnghar, K. Behnia, M. A. Pelleymounter, W. N. Washburn and W. R. Ewing, Bioorg. Med. Chem. Lett., 2007, 17, 3978–3982 CrossRef CAS PubMed.
- T. Meng, J. Wang, H. Peng, G. Fang, M. Li, B. Xiong, X. Xie, Y. Zhang, X. Wang and J. Shen, Eur. J. Med. Chem., 2010, 45, 1133–1139 CrossRef CAS PubMed.
- H. Fan, E. Kotsikorou, A. F. Hoffman, H. T. Ravert, D. Holt, D. P. Hurst, C. R. Lupica, P. H. Reggio, R. F. Dannals and A. G. Horti, Eur. J. Med. Chem., 2009, 44, 593–608 CrossRef CAS PubMed.
- R. L. Dow, J. R. Hadcock, D. O. Scott, S. R. Schneider, E. S. Paight, P. A. Iredale, P. A. Carpino, D. A. Griffith, M. Hammond and P. DaSilva-Jardine, Bioorg. Med. Chem. Lett., 2009, 19, 5351–5354 CrossRef CAS PubMed.
- R. L. Dow, P. A. Carpino, D. Gautreau, J. R. Hadcock, P. A. Iredale, D. Kelly-Sullivan, J. S. Lizano, R. E. O'Connor, S. R. Schneider, D. O. Scott and K. M. Ward, ACS Med. Chem. Lett., 2012, 3, 397–401 CrossRef CAS PubMed.
- J. H. M. Lange, H. H. van Stuivenberg, W. Veerman, H. C. Wals, B. Stork, H. K. A. C. Coolen, A. C. McCreary, T. J. P. Adolfs and C. G. Kruse, Bioorg. Med. Chem. Lett., 2005, 15, 4794–4798 CrossRef CAS PubMed.
- F. Ooms, J. Wouters, O. Oscari, T. Happaerts, G. Bouchard, P. Carrupt, B. Testa and D. M. Lambert, J. Med. Chem., 2002, 45, 1748–1756 CrossRef CAS PubMed.
- A. Fulp, K. Bortoff, H. Seltzman, Y. Zhang, J. Mathews, T. S. RFennell and R. Maitra, J. Med. Chem., 2012, 55, 2820–2834 CrossRef CAS PubMed.
- A. Fulp, K. Bortoff, Y. Zhang, R. Snyder, T. Fennell, J. A. Marusich, J. L. Wiley, H. Seltzman and R. Maitra, J. Med. Chem., 2013, 56, 8066–8072 CrossRef CAS PubMed.
- G. G. Muccioli, D. Martin, G. K. E. Scriba, W. Poppitz, J. H. Poupaert, J. Wouters and D. M. Lambert, J. Med. Chem., 2005, 48, 2509–2517 CrossRef CAS PubMed.
- Maestro 9.4, Schrödinger, LLC, New York, NY, 2013.
- R. Kristama, V. Parmara and V. N. Viswanadhana, J. Mol. Graphics Modell., 2013, 45, 157–172 CrossRef PubMed.
- P. R. Murumkar, M. K. Sharma, A. C. Shinde and K. G. Bothara, Med. Chem. Res., 2013, 22, 4192–4201 CrossRef CAS.
- LigPrep, version 2.3, Schrödinger, LLC, New York, NY, 2009.
- S. Kalva, E. R. A. Singam, V. Rajapandian, L. M. Saleena and V. Subramanian, J. Mol. Graphics Modell., 2014, 49, 25–37 CrossRef CAS PubMed.
- P. J. Therese, D. Manvar, S. Kondepudi, M. B. Battu, D. Sriram, A. Basu, P. Yogeeswari and N. K. Basu, J. Chem. Inf. Model., 2014, 54, 539–552 CrossRef CAS PubMed.
- M. Kaur, A. Kumari, M. S. Bahia and O. Silakari, J. Mol. Graphics Modell., 2013, 39, 165–175 CrossRef CAS PubMed.
- P. R. Murumkar, V. P. Zambre and M. R. Yadav, J. Comput.-Aided Mol. Des., 2010, 24, 143–156 CrossRef CAS PubMed.
- G. Li, L. Yanga, Y. Xua, W. Wanga, L. L. Li and S. Yanga, J. Mol. Graphics Modell., 2013, 44, 278–285 CrossRef CAS PubMed.
- Q. Huang, L. L. Li and S. Y. Yang, J. Mol. Graphics Modell., 2010, 28, 775–787 CrossRef CAS PubMed.
- R. D. Cramer, J. D. Bunce and D. E. Patterson, Quant. Struct.-Act. Relat., 1988, 7, 18–25 CrossRef.
- S. Kamath and J. K. Buolamwini, J. Med. Chem., 2003, 46, 4657–4668 CrossRef CAS PubMed.
- W. J. Dunn, S. Wold, V. Edlund and S. Helberg, Quant. Struct.-Act. Relat., 1984, 3, 131–137 CrossRef CAS.
- P. P. Roy, S. Paul, I. Mitra and K. Roy, Molecules, 2009, 14, 1660–1701 CrossRef CAS PubMed.
- P. P. Roy and K. Roy, QSAR Comb. Sci., 2008, 27, 302–313 CAS.
- S. Saha and G. P. S. Raghava, Nucleic Acids Res., 2006, 34, W202–W209 CrossRef CAS PubMed.
- www.zinc.docking.org, accessed 12 July 2014.
- Glide version 5.5, Maestro 9.0, Schrödinger, LLC, New York, NY, 2009.
- G. Kuang, G. Hu, X. Sun, W. Li, G. Liu and Y. Tang, J. Mol. Model., 2012, 18, 3831–3845 CrossRef CAS PubMed.
- P. R. Murumkar, M. K. Sharma, R. Giridhar and M. R. Yadav, Med. Chem. Res., 2015, 24, 226–244 CrossRef CAS.
- K. Roy, S. Kar and P. Ambure, Chemom. Intell. Lab. Syst., 2015, 145, 22–29 CrossRef CAS.
- QikProp, version 3.2, Schrödinger, LLC, New York, NY, 2009.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20612j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.