DOI:
10.1039/C5RA20443G
(Paper)
RSC Adv., 2016,
6, 3341-3349
Dicarboxylate ligand-modulated assembly of new luminescent zinc(II) coordination polymers with in situ formed tetrazole ligands: an experimental and theoretical study†
Received
2nd October 2015
, Accepted 16th December 2015
First published on 18th December 2015
Abstract
In situ hydrothermal reactions of Zn(NO3)2·6H2O, NaN3, and CPI with H2DPA or H2PYDC gave rise to two new zinc(II) coordination polymers, [Zn3(IPT)2(DPA)2] (1) and [Zn3(IPT)2(PYDC)2]·2H2O (2) (CPI = 1-(4-cyanophenyl)imidazole, HIPT = 5-[4-(imidazol-1-yl)phenyl]tetrazole, H2DPA = [1,1′-biphenyl]-2,2′-dicarboxylic acid and H2PYDC = pyridine-2,4-dicarboxylic acid), which were characterized by elemental analysis, IR, TGA and single crystal X-ray diffraction. Complex 1 features a new two-dimensional metal–organic layer with (3,6)-connected kgd topology based on cationic [Zn3(DPA)2]2+ clusters and anionic IPT ligands and further forms a three-dimensional supramolecular architecture via weak C–H⋯O hydrogen-bond interactions. However, complex 2 exhibits a three-dimensional framework formed from cationic [Zn3(PYDC)2]2+ chains and anionic IPT ligands, displaying a rare tetranodal (3,3,4,4)-connected net with 3,3,4,4T76 topology. Interestingly, the IPT ligands display different conformations and coordination modes in 1 and 2 due to the effect of the ancillary dicarboxylate co-ligands. All these data indicated that the nature of the ancillary dicarboxylate co-ligands plays an important role in diversifying the resulting architectures. The luminescence properties and thermal stabilities of the two complexes have also been investigated. Finally, high level theoretical calculations were performed to examine the conformations and coordination modes of the IPT ligands as well as the photophysical properties of the two complexes.
1. Introduction
Currently, there is much interest in the rational design and construction of coordination polymers as crystal functional materials that combine both inorganic and organic building blocks into a uniform architecture. This is because these materials have intriguing and diverse molecular topologies and crystal packing motifs, as well as potential applications in varied areas such as optics, electricity, magnetism, and catalysis.1 As is well known, the inherent nature of the bridging organic ligands, such as their coordination capabilities, coordination modes, conformations, substituents and flexibility/rigidity, can directly affect the structures and properties of the resulting complexes; thus, the selection of bridging organic ligands is very crucial in the assembly of these interesting coordination complexes.2 Recently, neutral N-containing bridging organic ligands as assembly blocks have attracted significant interest for their excellent coordination capacities and their potential applications in advanced materials.3 Especially, Sharpless et al. described a novel, safe and effective strategy for the preparation of 5-substituted-1H-tetrazoles through the [2 + 3] cycloaddition reaction of an azide salt and an organic nitrile in the presence of a Lewis acid catalyst;4 since then, a large number of tetrazole-based metal coordination complexes bearing interesting architectures and properties have been reported.5 To date, exploration of the influence of auxiliary carboxylate ligands, which play an important role in the field of coordination chemistry,6 on the in situ synthesis and self-assembly of tetrazole-based metal complexes has received less consideration.7 Currently, HIPT, an excellent organic bifunctional building block, which combines one imidazole group and one tetrazole group, has been investigated in the construction of metal complexes;8 all of these investigations have demonstrated that the HIPT ligand is an excellent multidentate ligand that possesses good coordination capacity and various coordinated modes, yet it has not been well shown to be useful in the synthesis of metal complexes, owing to many factors that influence the assembly process. Given the abovementioned consideration and as an extension of our previous studies on the in situ synthesis of tetrazole-based metal complexes,9 we have focused on exploring IPT-based metal coordination complexes generated in the presence of auxiliary H2DPA or H2PYDC ligands in the present study. Fortunately, two new coordination polymers, [Zn3(IPT)2(DPA)2] (1) and [Zn3(IPT)2(PYDC)2]·2H2O (2), have been successfully synthesized in situ under hydrothermal conditions. Both complexes exhibit different intriguing architectures and photoluminescence properties. Moreover, an extended computational analysis was carried out to better explore the conformations and coordination modes of the IPT ligand as well as the photophysical properties of the two complexes. We hope that this experimental and theoretical study will aid further exploration of the effects of tetrazole-based metal coordination complexes as auxiliary carboxylic ligands.
2. Experimental section
2.1. Materials and general methods
All starting chemicals for the syntheses were of analytical reagent grade and were used as received. The CPI was prepared according to a reported procedure.10 Elemental analyses (C, H and N) were performed on a Vario EL III analyzer. Infrared spectra (KBr pellets, 4000–400 cm−1) were acquired on a Nicolet AVATAR-360 spectrophotometer. Thermal analyses were determined on a Netzsch STA-409PC analyzer with a heating rate of 10 °C min−1 in flowing N2. The solid-state emission spectra were obtained at room temperature using a Hitachi F-4500 fluorescence spectrophotometer.
2.2. Synthesis of the complexes 1 and 2
[Zn3(IPT)2(DPA)2] (1). An aqueous solution (12 mL) of Zn(NO3)2·6H2O (59.2 mg, 0.2 mmol), NaN3 (13.2 mg, 0.2 mmol), CPI (33.8 mg, 0.2 mmol) and H2DPA (24.2 mg, 0.1 mmol) was sealed in a 25 mL Teflon-lined stainless steel container and heated at 180 °C for 3 days under autogenous pressure. Colorless rod crystals of 1 were collected with a yield of 51% based on H2DPA. Calcd (%) for C48H30N12O8Zn3 (1098.95): C, 52.46; H, 2.75; N, 15.29. Found (%): C, 52.38; H, 2.84; N, 15.43. IR (KBr/pellet)/cm−1: 3117(m), 3105(m), 2924(m), 1637(s), 1613(s), 1578(s), 1553(s), 1515(s), 1464(m), 1429(m), 1401(m), 1360(s), 1309(m), 1268(m), 1156(w), 1128(m), 1064(m), 1010(w), 956(w), 844(m), 759(m), 732(w), 685(w), 653(w), 617(w), 595(w), 559(w), 491(w), 467(w).
[Zn3(IPT)2(PYDC)2]·2H2O (2). The preparation of 2 was similar to that of 1 except that H2PYDC (16.7 mg, 0.1 mmol) was used instead of H2DPA. Colorless block crystals of 2 were obtained with a yield of 43% based on H2PYDC. Calcd (%) for C34H24N14O10Zn3 (984.78): C, 41.47; H, 2.46; N, 19.91. Found (%): C, 41.42; H, 2.51; N, 19.85. IR (KBr/pellet)/cm−1: 3138(m), 1616(s), 1548(m), 1515(w), 1454(w), 1430(w), 1390(s), 1306(w), 1265(w), 1134(w), 1066(w), 1029(w), 1009(w), 64(w), 925(w), 887(w), 823(w), 778(w), 731(m), 692(m), 644(w) 542(w), 497(w).
2.3. X-ray crystallography
Diffraction data collection for 1 and 2 was performed on a Bruker Smart CCD diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at 296(2) and 293(2) K, respectively. The structures of 1 and 2 were solved via direct method and refined on F2 via the full-matrix least-squares technique. All non-hydrogen atoms were refined with the anisotropic displacement parameters, whereas the hydrogen atoms of H2O were obtained via a difference Fourier map, and the hydrogen atoms of the carbon atoms were generated geometrically. All calculations were carried out with the SHELXL-97 program package.11 The crystallographic details and selected geometrical parameters for 1 and 2 can be found in Tables 1 and 2, respectively.
Table 1 Crystallographic data and structure refinement summary for 1 and 2
| R1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = |∑w(|Fo|2 − |Fc|2)|/∑|w(Fo)2|1/2, w = 1/[σ2(Fo2) + (aP)2 + bP]. P = (Fo2 + 2Fc2)/3. |
| Complexes |
1 |
2 |
| Formula |
C48H30N12O8Zn3 |
C34H24N14O10Zn3 |
| Formula weight |
1098.95 |
984.78 |
| T (K) |
296(2) |
293(2) |
| Crystal system |
Monoclinic |
Triclinic |
| Space group |
C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) |
19.7484(11) |
8.43950(10) |
| b (Å) |
7.6185(4) |
10.4158(2) |
| c (Å) |
30.3815(17) |
11.11610(10) |
| α (°) |
90 |
104.4540(10) |
| β (°) |
105.814(5) |
102.4520(10) |
| γ (°) |
90 |
94.8650(10) |
| V (Å3) |
4398.0(4) |
913.98(2) |
| Z |
4 |
1 |
| Dcalc. (g cm−3) |
1.660 |
1.789 |
| μ (mm−1) |
1.696 |
2.033 |
| F(000) |
2224 |
496 |
| θ range (°) |
1.39–25.05 |
1.95–25.05 |
| Reflns. collected |
19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 252 252 |
10191 |
| Rint |
0.1207 |
0.0604 |
| Data/restraints/parameters |
3632/0/321 |
3224/3/285 |
| Goodness-of-fit on F2 |
1.008 |
1.003 |
| R1 [I > 2σ(I)]a |
0.0426 |
0.0317 |
| wR2 [I > 2σ(I)]b |
0.1210 |
0.0643 |
| R1 (all data)a |
0.0592 |
0.0424 |
| wR2 (all data)b |
0.1340 |
0.0667 |
| Largest diff. peak and hole (e A−3) |
1.156 and −0.819 |
0.396 and −0.324 |
Table 2 Selected bond lengths (Å) and bond angles (°) for 1 and 2a
| Symmetry transformations used to generate equivalent atoms: #1 −x + 1/2, −y − 1/2, −z; #2 −x + 1, y, −z + 1/2; #3 x + 1/2, −y + 1/2, z + 1/2; #4 −x + 1/2, −y + 1/2, −z; #5 −x, −y, −z + 1; #6 x, y − 1, z; #7 −x + 1, −y, −z + 2; #8 x, y − 1, z + 1; #9 −x + 1, −y + 1, −z + 1. |
| [Zn3(IPT)2(DPA)2] (1) |
| Zn(1)–O(2) |
1.909(3) |
Zn(2)–O(4) |
1.953(3) |
| Zn(1)–O(3) |
1.958(3) |
Zn(2)–O(4)#2 |
1.953(3) |
| Zn(1)–N(1) |
1.998(3) |
Zn(2)–N(4)#3 |
1.993(3) |
| Zn(1)–N(6)#1 |
2.008(3) |
Zn(2)–N(4)#4 |
1.993(3) |
| O(2)–Zn(1)–O(3) |
126.90(12) |
O(4)#2–Zn(2)–O(4) |
110.79(17) |
| O(2)–Zn(1)–N(1) |
110.12(13) |
O(4)#2–Zn(2)–N(4)#3 |
113.73(14) |
| O(3)–Zn(1)–N(1) |
102.28(12) |
O(4)–Zn(2)–N(4)#3 |
107.35(14) |
| O(2)–Zn(1)–N(6)#1 |
104.01(13) |
O(4)#2–Zn(2)–N(4)#4 |
107.35(14) |
| O(3)–Zn(1)–N(6)#1 |
106.15(13) |
O(4)–Zn(2)–N(4)#4 |
113.73(14) |
| N(1)–Zn(1)–N(6)#1 |
105.83(14) |
N(4)#3–Zn(2)–N(4)#4 |
103.8(2) |
|
| [Zn3(IPT)2(PYDC)2]·2H2O (2) |
| Zn(1)–O(1) |
1.921(2) |
Zn(2)–O(3)#7 |
2.094(2) |
| Zn(1)–O(4)#5 |
1.951(2) |
Zn(2)–N(1) |
2.128(2) |
| Zn(1)–N(2) |
1.980(2) |
Zn(2)–N(1)#7 |
2.128(2) |
| Zn(1)–N(7)#6 |
2.019(2) |
Zn(2)–N(4)#8 |
2.215(2) |
| Zn(2)–O(3) |
2.094(2) |
Zn(2)–N(4)#9 |
2.215(2) |
| O(1)–Zn(1)–O(4)#5 |
91.77(9) |
N(1)–Zn(2)–N(1)#7 |
180.000(1) |
| O(1)–Zn(1)–N(2) |
125.11(10) |
O(3)–Zn(2)–N(4)#8 |
91.65(8) |
| O(4)#5–Zn(1)–N(2) |
116.57(9) |
O(3)#7–Zn(2)–N(4)#8 |
88.35(8) |
| O(1)–Zn(1)–N(7)#6 |
111.13(9) |
N(1)–Zn(2)–N(4)#8 |
87.15(8) |
| O(4)#5–Zn(1)–N(7)#6 |
109.38(9) |
N(1)#7–Zn(2)–N(4)#8 |
92.85(8) |
| N(2)–Zn(1)–N(7)#6 |
102.51(9) |
O(3)–Zn(2)–N(4)#9 |
88.35(8) |
| O(3)–Zn(2)–O(3)#7 |
180.00(8) |
O(3)#7–Zn(2)–N(4)#9 |
91.65(8) |
| O(3)–Zn(2)–N(1) |
79.77(8) |
N(1)–Zn(2)–N(4)#9 |
92.85(8) |
| O(3)#7–Zn(2)–N(1) |
100.23(8) |
N(1)#7–Zn(2)–N(4)#9 |
87.15(8) |
| O(3)–Zn(2)–N(1)#7 |
100.23(8) |
N(4)#8–Zn(2)–N(4)#9 |
180.000(1) |
| O(3)#7–Zn(2)–N(1)#7 |
79.77(8) |
|
|
2.4. Computational details
All computations were performed with the Gaussian09 D.01 software package12 using the density functional B3LYP method13 and the standard 6-311++G(d, p) basis set.14 To illustrate the different conformations and coordination modes of the IPT ligands in 1 and 2, density functional theory (DFT) computations were carried out on the IPT ligands. Based on the optimized geometric structures of the IPT ligands, potential energy surface (PES) scans were computed via performing anticlockwise rotations around the C–N and C–C single bonds between the imidazole/tetrazole ring and phenyl ring with fully unrelaxed geometry at every other step of the scan. Electrostatic surface potentials, V(r), and average local ionization energies, Ī(r), were calculated using the Multiwfn program15 based on the minor calculational models (Fig. 3) selected from the crystal structure measurements. In addition, to interpret the photoluminescence properties of 1 and 2, we performed time-dependent DFT (TD-DFT) calculations at the B3LYP/6-311++G(d, p) level on the selected minor models. At the same time, the natural transition orbitals16 (NTOs) for the excited states, which were derived from the calculated transition densities, were further used to explore the nature of the excited states involved in the photophysical processes.
3. Results and discussion
3.1. Synthesis and spectral characterization
The hydrothermal method, which plays a prominent role in the assembly of novel functional materials with diverse structures and also alters reactions from the kinetic to the thermodynamic domain, was adopted to facilely obtain new coordination polymers in the present study. The bifunctional HIPT ligands in complexes 1 and 2 were produced via an in situ [2 + 3] cycloaddition reaction between NaN3 and CPI in the presence of Zn2+ ion as a Lewis acid catalyst. Especially, the dihedral angles between the imidazole/tetrazole rings and their corresponding phenyl rings can freely vary from 0° to 180° due to the different reaction conditions. The flexible H2DPA and rigid H2PYDC ligands, which have previously been employed as excellent building blocks in the construction of coordination polymers, were selected as the auxiliary dicarboxylate co-ligands due to their different geometric structures, polarities and coordinated modes, which enable the facile construction of coordination polymers bearing IPT ligands with different conformations. Moreover, 1 and 2 were not soluble in water and common organic solvents and were air-stable. In the FT-IR spectra, the absorption bands at 3117, 3105 and 2924 cm−1 for 1 and 3138 cm−1 for 2 are attributed to the aromatic C–H bond stretching vibrations. The absence of the characteristic absorption peaks of carboxyl groups at about 1700 cm−1 and cyano groups at about 2200 cm−1 suggests that the carboxyl groups were completely deprotonated and that the cyano group of CPI was converted into a tetrazolyl group, with several typical tetrazolyl group peaks observed at 1464 cm−1 (1), 1429 cm−1 (1), 1454 cm−1 (2) and 1430 cm−1 (2). In addition, the strong momentous shift of the absorption bands of the carboxyl groups at 1637 cm−1 (1) and 1616 cm−1 (2) can be assigned to the metal coordination of the carboxyl groups. The abovementioned attributions of the IR spectra of 1 and 2 coincide with the results of the single crystal X-ray analyses.
3.2. Description of crystal structures
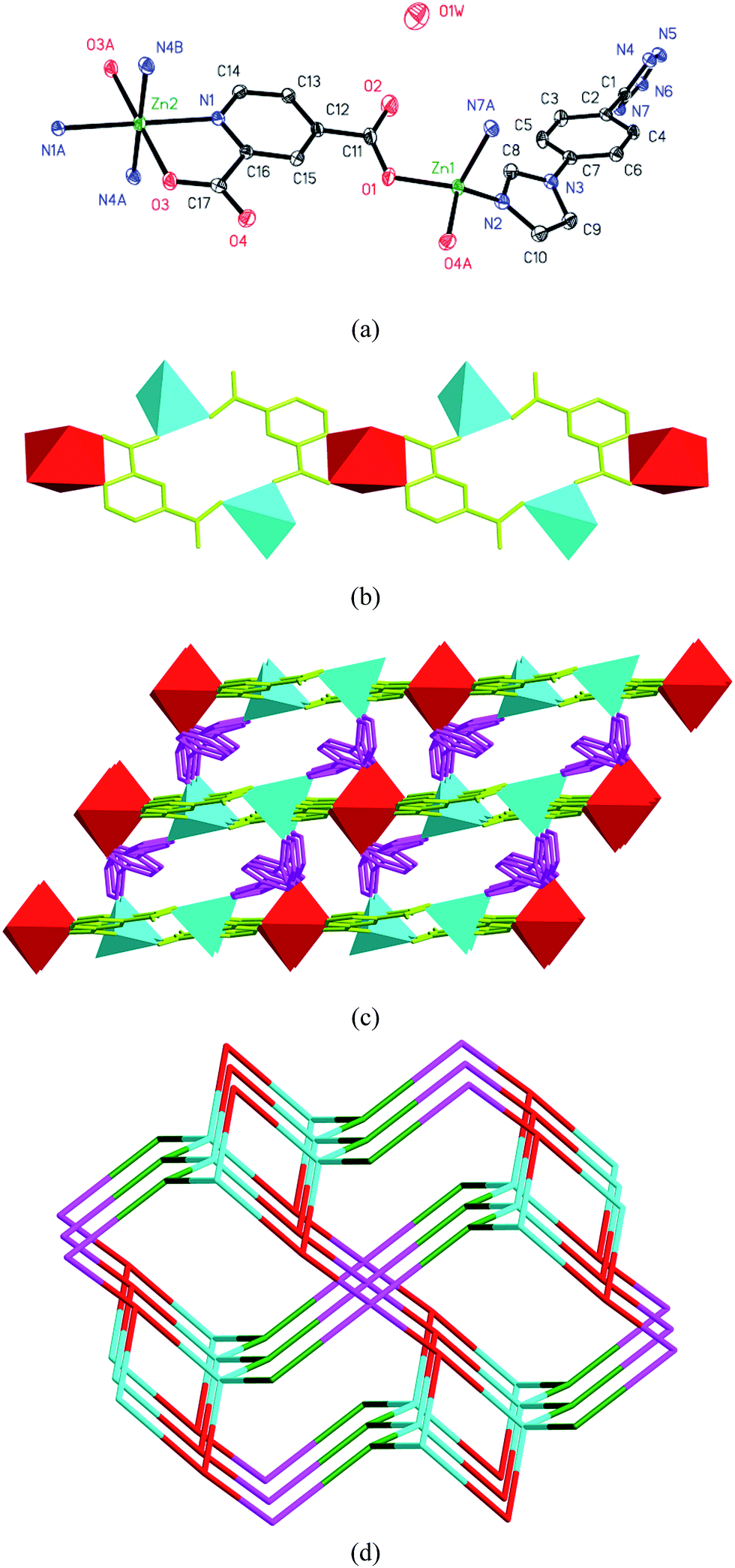
Structure of [Zn3(IPT)2(DPA)2] (1). Complex 1 crystallizes in the monoclinic space group C2/c; its asymmetric unit consists of one and a half zinc(II) atoms, one DPA ligand and one IPT ligand. As shown in Fig. 1a, the Zn1 center, which shows a slightly distorted tetrahedral geometry, is four coordinated via two carboxylate oxygen atoms (Zn–O = 1.909(3) and 1.958(3) Å) from one μ2-bridge DPA ligand (Scheme 1), as well as one tetrazole nitrogen atom (Zn–N = 2.008(3) Å) and one imidazole nitrogen atom (Zn–N = 1.998(3) Å) from two different μ3-bridge IPT ligands (IPT-A, Scheme 1). The angles around the Zn1 atom range from 102.28(12)° to 126.90(12)°. Comparably, the Zn2 atom is located on a 2-fold axis in the b-axis direction and is also four coordinated via two carboxylate oxygen atoms (Zn–O = 1.953(3) Å) from two different μ2-bridge DPA ligands, as well as two tetrazole nitrogen atoms (Zn–N = 1.993(3) Å) from two different μ3-bridge IPT ligands. The angles around the Zn2 atom range from 103.8(2)° to 113.73(14)°. Moreover, the two adjacent Zn1 atoms and one Zn2 atom are bridged by two DPA ligands to generate a homotrinuclear zinc(II) cationic cluster, [Zn3(DPA)2]2+. Interestingly, the [Zn3(DPA)2]2+ clusters sit on a 2-fold axis in the b-axis direction, resulting in a perfect isosceles triangle of zinc(II) atoms with Zn1⋯Zn2 = 4.7219(2) Å and Zn1⋯Zn1 = 7.2790(4) Å; the vertex angle of Zn1⋯Zn2⋯Zn1 is 100.847(1)°. Furthermore, the trinuclear [Zn3(DPA)2]2+ clusters are connected via six IPT ligands to generate an infinite two-dimensional distinct metal–organic layer parallel to the (10−1) plane (Fig. 1b). Notably, the Zn2 atoms in the abovementioned two-dimensional layer are distributed in a completely local plane, whereas the Zn1 atoms are distributed in two different parallel planes, with a separated interlayer distance of 1.9045 Å between the (Zn1)n/2 and (Zn2)n planes; moreover, the distance between the two (Zn1)n/2 planes is 3.8090 Å. From the topological view, each [Zn3(DPA)2]2+ cluster is considered as a six-connected node, whereas each μ3-bridge IPT ligand is considered as a three-connected node; thus, this two-dimensional metal–organic layer can be classified as a rare binodal (3,6)-connected topological net with the point (Schläfli) symbol of (43)2(46·66·83), validated via TOPOS,17 showing a typical kgd topology (Fig. 1c). Furthermore, through weak C–H⋯O hydrogen-bond interactions (C5–H5⋯O1 [−1/2 + x, 1/2 + y, z] = 3.196(6) Å/144°), the adjacent two-dimensional layers are further stacked in a face-to-face fashion along the [1−10] direction into a three-dimensional supramolecular framework with an adjacent interlayer distance of 9.192 Å, (Zn2)n planes (Fig. 1c and S1†).
 |
| | Fig. 1 (a) Coordination environments of the zinc(II) centers in 1 with the ellipsoids drawn at the 30% probability level; all hydrogen atoms are omitted for clarity. (b) Polyhedral representation of the two-dimensional layer-like structure parallel to the (10−1) plane in 1. (c) Schematic of the three-dimensional supramolecular stacking structure in 1. | |
 |
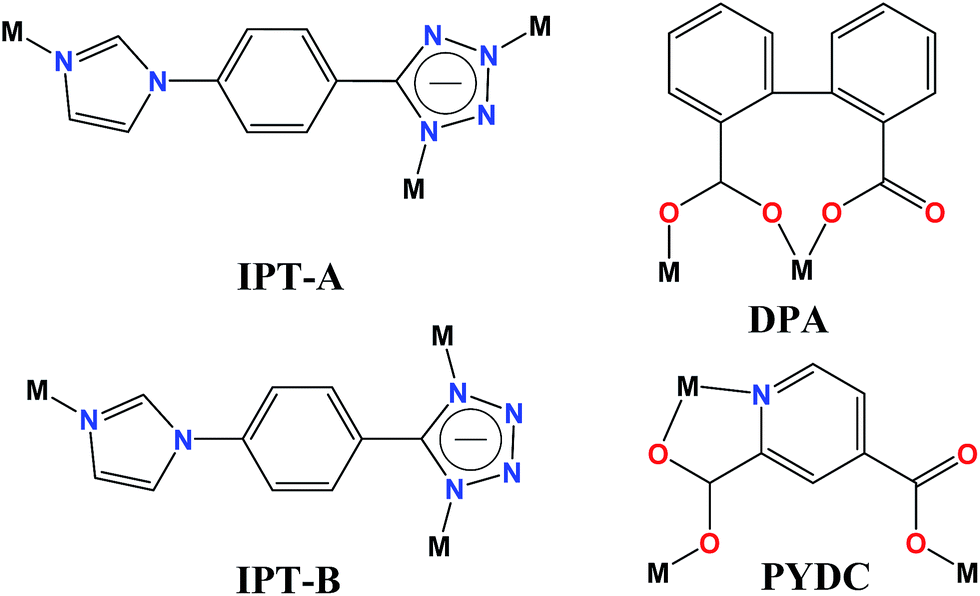
| | Scheme 1 Coordination modes of the IPT, DPA and PYDC anions. | |
Structure of [Zn3(IPT)2(PYDC)2]·2H2O (2). When the rigid auxiliary dicarboxylate ligand H2PYDC was used instead of the flexible dicarboxylate ligand H2DPA under similar reaction conditions to those used to obtain compound 1, we obtained a completely different framework for compound 2. Complex 2 crystallizes in the triclinic space group P; its asymmetric unit consists of one and a half zinc(II) atoms, one PYDC ligand, one IPT ligand and one uncoordinated water molecule, as shown in Fig. 2a. Interestingly, two independent zinc(II) atoms present different coordination environments. The Zn1 atom, which presents a slightly distorted tetrahedral geometry, is four coordinated via two carboxylate oxygen atoms (Zn–O = 1.921(2) and 1.951(2) Å) from one μ3-bridge PYDC ligand (Scheme 1) and one tetrazole nitrogen atom (Zn–N = 2.019(2) Å) as well as one imidazole nitrogen atom (Zn–N = 1.980(2) Å) from two different μ3-bridge IPT ligands (IPT-B, Scheme 1). The angles around the Zn1 atom range from 91.77(9)° to 125.11(10)°. Comparably, the Zn2 atom with an N4O2 donor set sits on a crystallographically inverted center and is six coordinated via two carboxylate oxygen atoms (Zn–O = 2.094(2) Å) from two different μ3-bridge PYDC ligands, two tetrazole nitrogen atoms (Zn–N = 2.215(2) Å) from two different μ3-bridge IPT ligands, and two pyridine nitrogen atoms from two different μ3-bridge PYDC ligands, resulting in a distorted octahedral tetrahedral geometry. The angles around the Zn2 atom range from 79.77(8)° to 180.000(1)°. Each μ3-bridge PYDC ligand bridges three adjacent zinc(II) atoms to form a one-dimensional infinite chain running along the [101] direction (Fig. 2b). The adjacent one-dimensional infinite chains are further joined by μ3-bridge IPT ligands to form a three-dimensional framework (Fig. 2c). Noticeably, from a topological point of view, the Zn1 and Zn2 atoms can be both considered as 4-connected nodes, and the PYDC and IPT ligands are both considered as 3-connected nodes; thus, this three-dimensional framework can be classified as a tetranodal (3,3,4,4)-connected network with the point (Schläfli) symbol of (4·82)2(83)2(4·85)2 (85·12), displaying a rare 3,3,4,4T76 topology (Fig. 2d). Moreover, PLATON analysis indicates that the whole structure contains a free void space of 914.0 Å3, excluding water molecules, which occupy approximately 6.6% of the whole cell volume. The uncoordinated water molecules are inserted in the free void space and are linked with the framework of complex 2 via O–H⋯O and C–H⋯O hydrogen-bond interactions (O1W–H1WB⋯O2 [1 − x, 1 − y, 1 − z] = 3.010(5) Å/139(5)°, O1W–H1WA⋯O2 = 2.875(5) Å/168(5)° and C8–H8⋯O1W [1 − x, 1 − y, 1 − z] = 3.305(5) Å/160°), which help to stabilize the complex framework (Fig. S2†).
 |
| | Fig. 2 (a) Coordination environments of the zinc(II) centers in 2 with the ellipsoids drawn at the 30% probability level; all hydrogen atoms are omitted for clarity. (b) Polyhedral representation of the one-dimensional infinite chain structure along the [101] direction in 2. (c) Schematic of the three-dimensional structure in 2. (d) Schematic of the (3,3,4,4)-connected three-dimensional topology net. | |
3.3. Effect of ancillary dicarboxylate co-ligands on structures
Organic carboxylates, which have been widely used to construct coordination polymers due to their excellent selectivity and various coordination modes, play a critical role in modulating the molecular structures of the resulting complexes. In the present study, the nature of the ancillary dicarboxylate ligands, such as their rigidity/flexibility, geometric structures, polarities and coordinated modes, is an important factor for the structural differences of the two obtained zinc(II) coordination polymers. To investigate the influence of dicarboxylate ligands on the complex architectures, two related dicarboxylate ligands, H2DPA and H2PYDC, were employed. For 1 and 2, all the reaction conditions were the same except for the ancillary dicarboxylate ligands. As a result, compound 1, which is based on the flexible dicarboxylate ligand H2DPA displays a two-dimensional distinct metal–organic layer with a typical kgd topology. Moreover, 2, generated from the rigid auxiliary dicarboxylate ligand H2PYDC, possesses a different three-dimensional framework displaying a rare 3,3,4,4T76 topology. Especially, owing to the influence of auxiliary dicarboxylate ligands, the in situ generated IPT ligands show different conformations and coordination modes in the two resulting complexes. In 1, the dihedral angles between the imidazole/tetrazole ring and the phenyl ring are 13.011° and 86.555°, compared to 35.634° and 62.020° in 2. The different conformations of the IPT ligands led to the different coordination modes IPT-A and IPT-B (Scheme 1). To represent the conformations and coordination modes of the IPT ligands in 1 and 2, DFT calculations were performed on the IPT ligands (IPT-A and IPT-B, Fig. 3) generated the crystal structures. The optimized stable geometric conformation of the IPT ligand is distinctly different from those of IPT-A and IPT-B. The dihedral angles between the imidazole/tetrazole ring and the phenyl ring are 41.459° and 1.916°, which can be assigned to the internal rotations of C–N and C–C single bonds between the imidazole/tetrazole ring and the phenyl ring. Furthermore, based on the optimized structure, the internal rotation PES was obtained via performing anticlockwise rotations around the C–N and C–C single bonds. The corresponding internal rotation angles (ω1 and ω2) values range from 0° to 180°, while all other geometric parameters are fully fixed. As shown in Fig. 4, four stable conformers and three transition states exist. Both conformers, IPT-A and IPT-B, are located on the unstable point in the PES. Notably, the calculated maximum internal rotation energy barrier is only 23.37 kJ mol−1, which is far lower than the energy value of 83.6 kJ mol−1 generated from the thermal motion of molecules at room temperature; thus, the thermal motion of the molecules far overshoots the maximum internal rotation energy barrier between the imidazole/tetrazole ring and the phenyl ring, and the IPT ligand can exist in different conformations under hydrothermal conditions at 180 °C. According to the abovementioned computational analysis, the presence of the conformers IPT-A and IPT-B in 1 and 2 should be completely attributed to the nature of the ancillary dicarboxylate co-ligands. To illustrate the different coordination modes of IPT ligand in the two resulting complexes, the molecular electrostatic potentials, V(r), and the average local ionization energies, Ī(r), which can represent fundamental determinants of atomic and molecular properties and reactive behavior, were computed via the Multiwfn program, based on the wave function obtained at the B3LYP/6-311++G(d, p) level. Fig. 5 presents the locations and magnitudes of the local minima values, VS, min(r) and ĪS, min(r), on the vdW surface of IPT-A and IPT-B, which are the reactive positions in terms of the electrophile. The IPT-A is seen to have five local minima; VS, min(r) and ĪS, min(r) are associated with the nitrogen atoms of the IPT ligand. In considering the data, VS, min(r) and ĪS, min(r), the strongly reactive positions of the tetrazole group are located at the N-1 and N-3 positions, respectively, and the reactive position of the imidazole group is located on the N-atom, which contains a lone pair of electrons. Thus, the IPT ligand exists in the IPT-A coordination mode in 1 (Scheme 1). As for IPT-B, four VS, min(r) and five ĪS, min(r) values associated with the nitrogen atoms were also obtained. Compared with IPT-A, the reactive position of the imidazole group is also located on the N-atom bearing a lone pair of electrons, while the strong reactive positions of the tetrazole group are located on the N-1 and N-4 positions, respectively. As a consequence, another coordination mode, IPT-B, was exhibited in 2, which is in good agreement with the experimental results. All these findings indicated that the ancillary organic carboxylate co-ligands play a crucial role in altering the nature of the primary ligands and determining the resulting architectures.
 |
| | Fig. 3 Calculated ground state models of IPT-A and IPT-B. | |
 |
| | Fig. 4 Calculated color-filled contour plot of the potential energy surface of the optimized IPT anions at the B3LYP/6-311++G(d, p) level. | |
 |
| | Fig. 5 Calculated molecular electrostatic potential (left) and average local ionization energy (right) on the vdW surface of IPT-A and IPT-B at the B3LYP/6-311++G(d, p) level (unit: kJ mol−1). | |
3.4. Thermal gravimetric analyses
To verify the thermal stabilities of 1 and 2, thermogravimetric analyses (TGA) were carried out under a N2 atmosphere with a heating rate of 10 °C min−1. As depicted in Fig. S3,† 1 showed no weight loss until the temperature reached 240 °C; after that temperature, the framework of 1 began to decompose. For 2, the first weight loss of 3.8% in the temperature range of 60–240 °C corresponds to the loss of two uncoordinated water molecules (calcd 3.66%). The rapid decomposition of the framework occurred at about 280 °C, which is attributed to the decomposition of the IPT and PYDC groups. Moreover, both complexes did not decompose completely until the temperature reached 800 °C. The results of the TG analysis show that both complexes have good thermal stabilities.
3.5. Photoluminescence properties
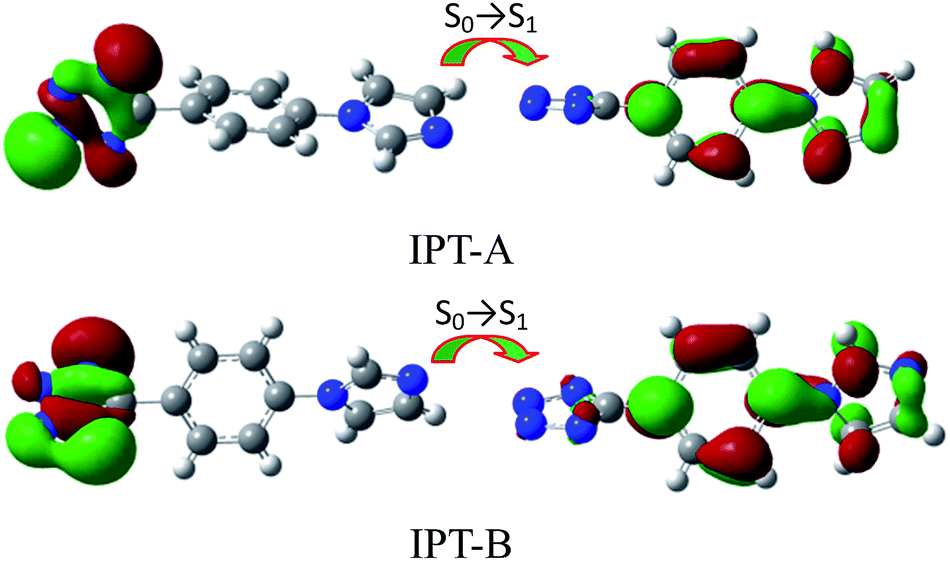
Luminescent zinc(II) complexes possessing closed d10 shells are superior potential candidates as valuable luminescent materials;18 thus, we probed the luminescence properties of 1 and 2 in the solid state at room temperature. As given in Fig. 6, weak emission peaks for 1 and 2 are observed at 368 nm (λex = 300 nm) and 326 nm (λex = 300 nm), respectively. Compared with the luminescence properties of the free HIPT at 362 nm (λex = 285 nm),19 the emission peaks of 1 and 2 present some red/blue shifts, which can be generally ascribed to the ligand-centered nature of the emission, due to the fact that the d10 Zn2+ ion is very difficult to oxidize and reduce.20 To further obtain the luminescence and electronic properties of 1 and 2, TD-DFT calculations were performed on the selected models (IPT-A and IPT-B). The TD-DFT computations indicate that the allowed lowest s0 → s1 vertical electronic transition can be completely dominated via the major HOMO → LUMO (97%, IPT-A, 76%, IPT-B), minor HOMO−1 → LUMO (15%, IPT-B) and HOMO−3 → LUMO (4%, IPT-B) contributions. Interestingly, the computational s0 → s1 vertical electronic transition energies of IPT-A and IPT-B are 3.49 eV (355.39 nm) and 3.94 eV (314.77 nm), respectively, which are in good agreement with the results of the abovementioned experimental exploration and also further indicate that the luminescence properties of 1 and 2 can be ascribed to the IPT-centered nature of their emissions. Furthermore, based on the computed transition density matrices, NTO calculations were also performed on the selected models; these calculations can demonstrate the most compact orbital picture of the electron transition between the ground and excited states. Fig. 7 exhibits the NTO pairs for the first excited singlet state of the selected models of IPT-A and IPT-B. The associated holes of IPT-A and IPT-B, which involve complete contributions from the HOMO for IPT-A and the HOMO, HOMO−1 and HOMO−3 for IPT-B, are all principally located on the tetrazole ring of the IPT ligand, whereas the corresponding associated particles of IPT-A and IPT-B from the LUMO contribution reside completely on the phenyl and imidazole rings of the IPT ligand. The computed largest NTO eigenvalues are all over 99% for the first excited singlet states, which means that the s0 → s1 transition states of IPT-A and IPT-B can both be represented quite well as a dominant excitation pair. This also means that the first excited singlet states for IPT-A and IPT-B have the character of IPT intra-ligand charge transfer transitions (ILCT). Therefore, the experimental weak low-energy emission bands for 1 and 2 in the solid state can all be assigned to ILCT mechanisms based on the abovementioned experimental and computational analyses.
 |
| | Fig. 6 The solid-state emission spectra of 1 and 2 at room temperature. | |
 |
| | Fig. 7 The dominant natural transition orbital pairs for the first excited singlet states of IPT-A and IPT-B (left, hole; right, particle). The associated eigenvalues are 0.99767 and 0.99669, respectively. | |
4. Conclusions
In conclusion, we successfully prepared two new zinc(II) coordination polymers containing rigid bifunctional tetrazole-based ligands and auxiliary organic carboxylate co-ligands via a simultaneous in situ self-assembly reaction under hydrothermal conditions. The two coordination polymers both present fascinating two-dimensional and three-dimensional architectures with rare topologies. The structural diversities of the two complexes indicate that the nature of the rigid IPT ligand and the auxiliary organic carboxylate co-ligands plays a subtle role in the construction of novel coordination architectures, and the successful syntheses of the two new complexes also show that this method is promising to obtain intriguing structures and properties via in situ ligand formation and modifying of the auxiliary ligands.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 20971004), the Natural Science Foundation of the Education Commission of Anhui Province, China (No. KJ2010A229) and the Outstanding Youth Foundation of the Education Commission of Anhui Province, China (No. 2010SQRL108ZD).
References
-
(a) R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093 CrossRef CAS;
(b) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 133 RSC;
(c) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed;
(d) H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed;
(e) D. Tanaka, A. Henke, K. Albrecht, M. Moeller, K. Nakagawa, S. Kitagawa and J. Groll, Nat. Chem., 2010, 2, 410 CrossRef CAS PubMed;
(f) S. C. Xiang, X. T. Wu, J. J. Zhang, R. B. Fu, S. M. Hu and X. D. Zhang, J. Am. Chem. Soc., 2005, 127, 16352 CrossRef CAS PubMed;
(g) D. Liu, Z. G. Ren, H. X. Li, J. P. Lang, N. Y. Li and B. F. Abrahams, Angew. Chem., Int. Ed., 2010, 49, 4767 CrossRef CAS PubMed;
(h) W. Walker, S. Grugeon, O. Mentre, S. Laruelle, J. M. Tarascon and F. Wudl, J. Am. Chem. Soc., 2010, 132, 6517 CrossRef CAS PubMed;
(i) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC;
(j) T. Y. Shvareva, S. Skanthakumar, L. Soderholm, A. Clearfield and T. E. Albrecht-Schmitt, Chem. Mater., 2007, 19, 132 CrossRef CAS.
-
(a) A. J. Blake, N. R. Brooks, N. R. Champness, M. Crew, A. Deveson, D. Fenske, D. H. Gregory, L. R. Hanton, P. Hubberstey and M. Schröder, Chem. Commun., 2001, 1432 RSC;
(b) Y. J. Qi, Y. H. Wang, C. W. Hu, M. H. Cao, L. Mao and E. B. Wang, Inorg. Chem., 2003, 42, 8519 CrossRef CAS PubMed;
(c) V. S. S. Kumar, F. C. Pigge and N. P. Rath, New J. Chem., 2004, 28, 1192 RSC;
(d) B. Li, G. Li, D. Liu, Y. Peng, X. Zhou, J. Hua, Z. Shi and S. Peng, CrystEngComm, 2011, 13, 1291 RSC.
-
(a) J. Xu, Z. R. Pan, T. W. Wang, Y. Z. Li, Z. J. Guo, S. R. Batten and H. G. Zheng, CrystEngComm, 2010, 12, 612 RSC;
(b) J. Y. Lu, Coord. Chem. Rev., 2003, 246, 327 CrossRef CAS;
(c) Y. Xu, P. K. Chen, Y. X. Che and J. M. Zheng, Eur. J. Inorg. Chem., 2010, 5478 CrossRef CAS;
(d) J. R. Li, D. J. Timmons and H. C. Zhou, J. Am. Chem. Soc., 2009, 131, 6368 CrossRef CAS PubMed;
(e) J. C. Jin, Y. N. Zhang, Y. Y. Wang, J. Q. Liu, Z. Dong and Q. Z. Shi, Chem.–Asian J., 2010, 5, 1611 CrossRef CAS PubMed;
(f) E. C. Yang, Z. Y. Liu, X. J. Shi, Q. Q. Liang and X. J. Zhao, Inorg. Chem., 2010, 49, 7969 CrossRef CAS PubMed.
-
(a) F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2003, 125, 9983 CrossRef CAS PubMed;
(b) Z. P. Demko and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2113 CrossRef CAS;
(c) Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945 CrossRef CAS PubMed.
-
(a) Q. Ye, X. S. Wang, H. Zhao and R. G. Xiong, Chem. Soc. Rev., 2005, 34, 208 RSC;
(b) H. Zhao, Z. R. Qu, H. Y. Ye and R. G. Xiong, Chem. Soc. Rev., 2008, 37, 84 RSC;
(c) B. Liu, Y. C. Qiu, G. Peng and H. Deng, CrystEngComm, 2010, 12, 270 RSC.
-
(a) R. Kieltyka, P. Englebienne, J. Fakhoury, C. Autexier, N. Moitessier and H. F. Sleiman, J. Am. Chem. Soc., 2008, 130, 10040 CrossRef CAS;
(b) D. Kong, J. Zon, J. McBee and A. Clearfield, Inorg. Chem., 2006, 45, 977 CrossRef CAS PubMed;
(c) H. A. Habib, J. Sanchiz and C. Janiak, Dalton Trans., 2008, 1734 RSC;
(d) X. Zhang, L. Hou, B. Liu, L. Cui, Y. Y. Wang and B. Wu, Cryst. Growth Des., 2013, 13, 3177 CrossRef CAS.
-
(a) M. Du, X. J. Jiang and X. J. Zhao, Chem. Commun., 2005, 5521 RSC;
(b) M. Du, X. J. Jiang and X. J. Zhao, Inorg. Chem., 2006, 45, 3998 CrossRef CAS PubMed;
(c) Y. Y. Lin, Y. B. Zhang, J. P. Zhang and X. M. Chen, Cryst. Growth Des., 2008, 8, 3673 CrossRef CAS;
(d) H. Ren, T. Y. Song, J. N. Xu, S. B. Jing, Y. Yu and P. Zhang, Cryst. Growth Des., 2009, 105 CrossRef CAS;
(e) G. Q. Lan, R. T. Dong, N. Q. Yu, L. M. Zhang, Y. Z. Ma, C. F. Yang and H. Deng, Inorg. Chem. Commun., 2012, 20, 295 CrossRef;
(f) L. Ma, Y. C. Qiu, G. Peng, J. B. Cai and H. Deng, Eur. J. Inorg. Chem., 2011, 23, 3446 CrossRef;
(g) L. Sun, L. Ma, J. B. Cai, L. Liang and H. Deng, CrystEngComm, 2012, 14, 890 RSC;
(h) Z. Y. Liu, X. J. Shi, E. C. Yang and X. J. Zhao, Inorg. Chem. Commun., 2010, 13, 1259 CrossRef CAS.
-
(a) D. Y. Ma, L. Qin, K. Lu, H. F. Guo and J. Q. Liu, Inorg. Chem. Commun., 2012, 24, 87 CrossRef CAS;
(b) J. Sun, D. Zhang, L. Wang, R. Zhang, J. Wang, Y. Zeng, J. Zhan, J. Xu and Y. Fan, J. Solid State Chem., 2013, 206, 286 CrossRef CAS;
(c) Z. Shi, J. Peng, Z. Zhang, X. Yu, K. Alimaje and X. Wang, Inorg. Chem. Commun., 2013, 33, 105 CrossRef CAS;
(d) H. W. Kuai, X. C. Cheng and X. H. Zhu, Inorg. Chem. Commun., 2012, 25, 43 CrossRef CAS;
(e) S. S. Hou, J. B. Tan, Z. y. Lian, D. W. Zeng, T. L. Huang, B. R. Huang, S. R. Zheng, J. Fan and W. G. Zhang, Inorg. Chem. Commun., 2014, 47, 112 CrossRef CAS;
(f) Z. Y. Shi, Z. Y. Zhang, J. Peng, X. Yu and X. Wang, CrystEngComm, 2013, 15, 7199 RSC;
(g) X. Wang, J. Peng, K. Alimaje and Z. Y. Shi, CrystEngComm, 2012, 14, 8509 RSC.
-
(a) R. Y. Huang, C. H. Zhu, X. J. Kong and H. Xu, Inorg. Chem. Commun., 2015, 61, 1 CrossRef CAS;
(b) R. Y. Huang, C. Xue, Z. Q. Wang, H. Xu, G. H. Wu and S. Y. Ye, Inorg. Chim. Acta, 2013, 405, 302 CrossRef CAS;
(c) X. Q. Zhao, C. Guo, R. Y. Huang, H. Xu, G. X. Liu and G. H. Wu, Chin. J. Struct. Chem., 2012, 10, 1501 Search PubMed;
(d) R. Y. Huang, K. Zhu, H. Chen, G. X. Liu and X. M. Ren, Chin. J. Inorg. Chem., 2009, 25, 162 CAS.
- A. Hatzidimitriou, A. Gourdon, J. Devillers, J. P. Launay, E. Mena and E. Mouyal, Inorg. Chem., 1996, 35, 2212 CrossRef CAS PubMed.
-
(a) G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, University of Göttingen, Germany, 1997 Search PubMed;
(b) G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
-
(a) A. D. Becke, Phys. Rev. A, 1998, 38, 3098 CrossRef;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CAS.
-
(a) S. Y. Liu, X. L. Qi, R. B. Lin, X. N. Cheng, P. Q. Liao, J. P. Zhang and X. M. Chen, Adv. Funct. Mater., 2014, 24, 5866 CrossRef CAS;
(b) F. Wu, J. Li, H. Tong, Z. Li, C. Adachi, A. Langlois, P. D. Harvey, L. Liu, W. Y. Wong, W. K. Wong and X. Zhu, J. Mater. Chem. C, 2015, 3, 138 RSC;
(c) Q. Xiao, J. Zheng, M. Li, S. Z. Zhan, J. H. Wang and D. Li, Inorg. Chem., 2014, 53, 11604 CrossRef CAS PubMed;
(d) F. Wu, H. Tong, Z. Li, W. Lei, L. Liu, W. Y. Wong, W. K. Wong and X. Zhu, Dalton Trans., 2014, 43, 12463 RSC.
- S. S. Hou, J. B. Tan, Z. Y. Lian, D. W. Zeng, T. L. Huang, B. R. Huang, S. R. Zheng, J. Fan and W. G. Zhang, Inorg. Chem. Commun., 2014, 47, 112 CrossRef CAS.
-
(a) Y. Yang, P. Du, J. F. Ma, W. Q. Kan, B. Liu and J. Yang, Cryst. Growth Des., 2011, 11, 5540 CrossRef CAS;
(b) F. Wang, X. Jing, B. Zheng, G. Li, G. Zeng, Q. Huo and Y. Liu, Cryst. Growth Des., 2013, 13, 3522 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1429190 and 1429191 contain the supplementary crystallographic data for 1 and 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra20443g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.