
Comparative study of dG affinity vs. DNA methylation modulating properties of side chain derivatives of procainamide: insight into its DNA hypomethylating effect†
Abstract
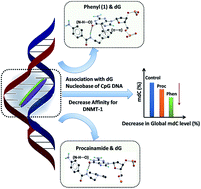
Procainamide derivatives have been synthesized to investigate the role of side chains in modulating the DNA methylation level in cancer cells and gain insight into its mechanism of action. The synthesized derivatives comprised of flexible (dimethyl), constrained (pyrrolidine, piperidine, morpholine) and planar aromatic (pyridine, phenyl) side chain motifs. The affinity of procainamide and its derivatives towards the deoxyguanosine (dG) base in neutral form has been assessed by performing Differential Pulse Voltammetry (DPV) under physiological conditions. Further, molecular docking with hemimethylated CpG rich DNA acquired from an active mDNMT-1-DNA (PDB ID-4DA4) crystal structure, reveals their preferential non-covalent interaction with dG nucleobase in the intercalation cavity of the minor groove. Differential affinity of the derivatives to dG base in neutral and bound forms (DNA) is correlated with their DNA methylation modulating properties at sub-lethal concentrations. Among all the derivatives, a compound with an aromatic phenyl side chain (1) has shown a highest binding affinity for dG nucleobase in neutral form as well as for partially denatured CpG rich DNA which is attributed to the formation of π⋯π stacking interaction in addition to N–H⋯O hydrogen bonding with the pyrimidine ring of dG base. It also shows the highest cytotoxicity and global hypomethylation at a sub-lethal level in the MCF-7 cancer cell line compared to other derivatives and procainamide. A docking study has also illustrated the plausible structural basis of DNA methylation modulating a property of procainamide. Strong association of procainamide with dG bases of partially denatured CpG rich DNA via H-bonding and other non-covalent interactions may alter the active topology of DNA required by the DNA-binding regulatory proteins (e.g. DNMT-1) which is validated by a DNMT-1 inhibition assay. This systematic investigation leads to a new potent alternative to procainamide being found and gives a plausible insight into the DNA hypomethylating effect of procainamide.
 Please wait while we load your content...
Please wait while we load your content...

