
Following Ramachandran: exit vector plots (EVP) as a tool to navigate chemical space covered by 3D bifunctional scaffolds. The case of cycloalkanes†
Oleksandr O. Grygorenko*a,
Pavlo Babenkoa,
Dmitry M. Volochnyukb,
Oleksii Raievskyicd and
Igor V. Komarova
aTaras Shevchenko National University of Kyiv, Volodymyrska Street 60, Kyiv 01601, Ukraine. E-mail: gregor@univ.kiev.ua
bInstitute of Organic Chemistry National Academy of Sciences of Ukraine, Murmanska Street 5, Kyiv 02094, Ukraine
cInstitute of Molecular Biology and Genetics National Academy of Sciences of Ukraine, Zabolotnogo Street 150, Kyiv 03680, Ukraine
dLife Chemicals, Life Chemicals Group, 1A Dixie Avenue, Niagara-on-the-Lake ON L0S 1J0, Canada
First published on 4th February 2016
Abstract
An approach to the analysis and visualization of chemical space covered by disubstituted scaffolds, which is based on exit vector plots (EVPs), is used for analysis of the simplest disubstituted cyclic cores – cycloalkanes – deposited in the Cambridge Structural Database (CSD). It is shown that four clearly defined regions are found in EVPs of the cycloalkanes, similar to those observed in Ramachandran plots for peptides. These results can be used for directed design of more complex scaffolds, classification of conformational space for the disubstituted scaffolds, rational scaffold replacement, or SAR studies.
Introduction
The concept of chemical space, i.e. descriptor space covered by all the possible molecules, is widely used to analyze compound libraries in combinatorial chemistry, drug discovery and many other areas.1–3 Due to the huge size of the chemical space, efficient strategies for its exploration and navigation,4–9 as well as visualization,10–12 are required. Molecular scaffolds are among the tools widely used nowadays for this purpose. Initially defined by Bemis and Murcko as combinations of ring systems and linkers connecting them,13 the scaffolds were widely used in medicinal chemistry and chemoinformatics to assess the diversity of compound libraries.14–24The well-established tendency in drug discovery is related to shifting towards three-dimensional, non-flattened scaffolds in the design of the compound libraries.25–29 A number of approaches to quantitative analysis of molecular “three-dimensionality” can be found in the literature, including the use of either very simple molecular descriptors such as fraction of sp3 carbon atoms (Fsp3)25 or more sophisticated (but still convenient) parameters like principal moments of inertia,30 plane of best fit,31 alpha shapes,32 ultrafast shape recognition (USR) descriptors,33 3D Zernike descriptors,34 harmonic pharma chemistry coefficient (HPCC),35 shape fingerprints,36 torsion fingerprints (TFD),37 radius of gyration and shadow indices.38 Most of these descriptors consider the overall shape of the molecule and do not count for relative orientation of particular functional groups mounted onto the central molecular scaffold. However, functional groups and other fragments connected to scaffolds play the central role in the drug–target interaction. Therefore it would be useful to have simple parameters allowing quantifying relative spatial orientation of the scaffold functionalities.
Torsion angles are the most obvious parameters which can be used to assess the relative orientation of the molecular fragments attached to the scaffold. One of the most prominent examples of using these parameters were well-known Ramachandran (φ–ψ) plots, introduced for description of peptide backbone conformation in 1960s40 (Fig. 1). These plots were convenient to use even with low calculation capacities available in those years, and they soon became a golden standard for discussing the three-dimensional structure of peptides and proteins. The use of torsion angles was extended for β- and other ω-peptides,41 and they were also evaluated for the purposes of medicinal chemistry.42–44
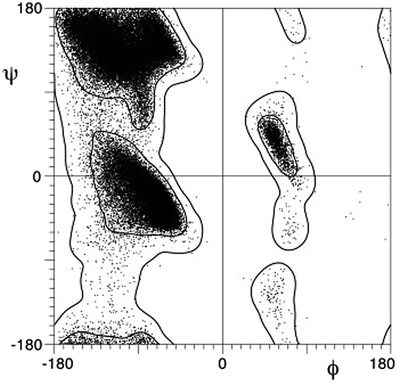 | ||
| Fig. 1 Ramachandran plot (reproduced from ref. 39). | ||
The major drawback of torsion angles as conformational descriptors of molecular scaffolds is the fact that multiple parameters are needed in general case; moreover, their number is varied for different scaffolds. Being inspired by the idea of simple visualization of molecular geometry provided by Ramachandran plots, we turned our attention to an alternative approach based on the so-called exit vectors, introduced for CAVEAT software in 1990s.45 Recently, we have used them for exploration of conformationally restricted diamines46–48 and other related molecules.49 In this approach, the functional groups mounted onto a scaffold are simulated by bound unit vectors. The variation points of the scaffold are used as the starting points of these vectors, whereas to define their directions, either the bonds to the substituents (for the carbon attachment points) or the bisectors of the corresponding valence angles (for the nitrogen attachment points, which are usually configurationally unstable) are used. In the case of bifunctional scaffolds, the relative orientation of the two exit vectors n1 and n2 can be described by four geometric parameters defined in Fig. 2 for 1,2-disubstituted cyclohexane: the distance between the variation points N1 and N2–r, the plane angles φ1 (between vectors n1 and N2N1) and φ2 (between n2 and N1N2), and the dihedral angle θ defined by vectors n1, N1N2 and n2.46–49 These parameters can be calculated easily from atomic coordinates and then used for the construction of Ramachandran-like plots (r–θ, θ–φ1/φ2, and φ1–φ2) and further analysis of the library distribution over their three-dimensional geometric properties.
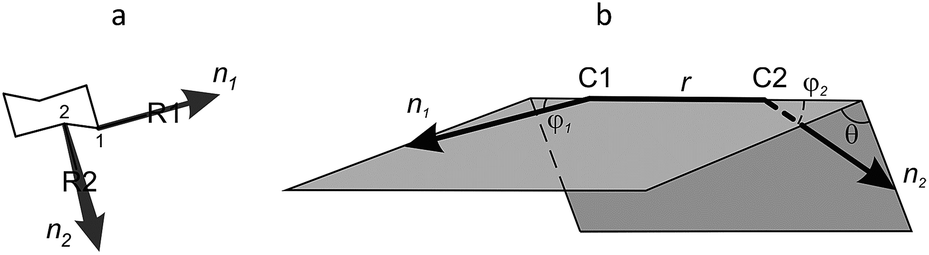 | ||
| Fig. 2 (a) Definition of vectors n1, n2 (1,2-disubstituted cyclohexane scaffold is used as an example). (b) Definition of geometric parameters r, φ1, φ2, and θ. | ||
Whereas r can be used as a measure of the scaffold size, the angles φ1, φ2 and θ define relative spatial orientation of the exit vectors – a property which is important to all areas of chemistry where intermolecular interactions are involved. For example, in drug design, this disposition is critical for the interaction of the fragments attached to the scaffold with the biological targets. The values of φ1, φ2 and θ angles can be used for estimation of the scaffold shape. Thus, for linear core (1,4-disubstituted benzene), φ1 = φ2 = 0° (θ is undefined) (Fig. 3). For non-linear flat molecules (such as 1,2-disubstituted benzenes), φ1 and φ2 have non-zero values, whereas the absolute value of θ is close to 0° or 180°. Finally, for many disubstituted cycloalkanes (e.g. 1,2-disubstituted cyclohexanes) and other non-flattened scaffolds, all the three angles are far from 0° (and 180°). Analysis of such cores in terms of r, φ1, φ2 and θ parameters might be especially useful for molecular design in various fields, in particular, for scaffold replacement approaches or discovery of chemotypes with previously unexplored molecular geometries.
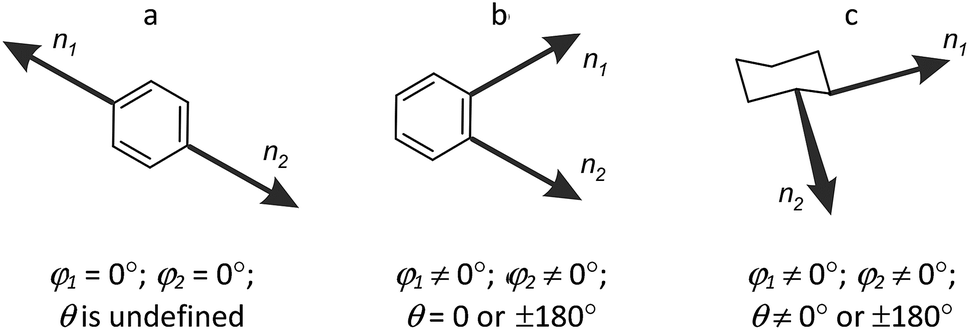 | ||
| Fig. 3 Parameters φ1, φ2, and θ as functions of the scaffold shape: (a) linear; (b) non-linear flat; (c) non-flattened. | ||
In this work, we report the construction and analysis of plots (“exit vector plots” (EVP)) in several coordinates, r–θ, θ–φ1/φ2, and φ1–φ2 for the simplest disubstituted cycloalkane scaffolds (cyclopropane, cyclobutane, cyclopentane, and cyclohexane), which is based on experimental data available from Cambridge Structural Database (CSD).50 Such use of the CSD has long been proven as an efficient instrument of structural, conformational and reactivity analysis.51 Fifty years since its establishment, the CSD has collected vast structural information (currently more than 750![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 entries). Each crystal structure deposited in the CSD was obtained for different and specific purposes (mainly for structure elucidation), but with more and more structural information available, it became possible to retrieve general structural relationships by statistical CSD analyses. Here, we propose a very simple EVP-based variant of such an analysis, which can be easily visualized. In our opinion, the results of such analysis should establish the basis for further use of the method for more complex systems. We expect that some relationships in the distribution of conformations over the EVP should be elucidated, similar to α or β regions of the Ramachandran diagram used for the explanation and prediction of typical peptide secondary structure elements.
000 entries). Each crystal structure deposited in the CSD was obtained for different and specific purposes (mainly for structure elucidation), but with more and more structural information available, it became possible to retrieve general structural relationships by statistical CSD analyses. Here, we propose a very simple EVP-based variant of such an analysis, which can be easily visualized. In our opinion, the results of such analysis should establish the basis for further use of the method for more complex systems. We expect that some relationships in the distribution of conformations over the EVP should be elucidated, similar to α or β regions of the Ramachandran diagram used for the explanation and prediction of typical peptide secondary structure elements.
It is known that conformational space of cycloalkanes has been studied thoroughly in previous works. Strauss–Pickett torsion angles,52 Cremer–Pople53 and Zefirov–Palyulin–Dashevskaya (ZPD)54 puckering parameters are the classical approaches to discuss cycloalkane conformations; some newer methods can be also mentioned such as triangular tessellation.55 However, these approaches can hardly be applied for general analysis of the whole chemical space of the disubstituted scaffolds. In this view, their major drawbacks are similar to those of usual torsion angles discussed above. All these approaches are limited by monocyclic ring systems; multiple parameter sets are needed in general case, and the number of these parameters is varied even for different cycloalkanes.
Results and discussion
Calculation of the geometric parameters
Atomic coordinates for the 1,2-disubstituted cycloalkanes were retrieved from CSD into CIF files using the substructure search. The data for each molecule were transferred into the database, and 1,2-disubstituted cycloalkane cores, as well as the variation points of exit vectors were identified using Open Babel software56 and in-house written Ruby script. The cores with undefined positions were removed from the consideration. The corresponding atomic coordinates for 2886 disubstituted cycloalkane cores thus obtained were used to calculate the geometric parameters r, φ1, φ2, and θ. Since the sign of θ angle is the only parameter which is affected by the chirality of the molecule, we have considered only absolute values of the θ, keeping in mind that corresponding negative values are also possible. In other words, we took only one enantiomer from the enantiomeric pairs for the analysis. Furthermore, for the choice of the carbon atom numbers of a cycloalkane scaffold was not arbitrary, we let φ1 > φ2. The calculated values were used to construct EVP (in r–θ, θ–φ1/φ2, and φ1–φ2 coordinates) for each of the ring systems discussed in this work and then – for the combined data set.Discussion of EVP for particular disubstituted cycloalkane rings
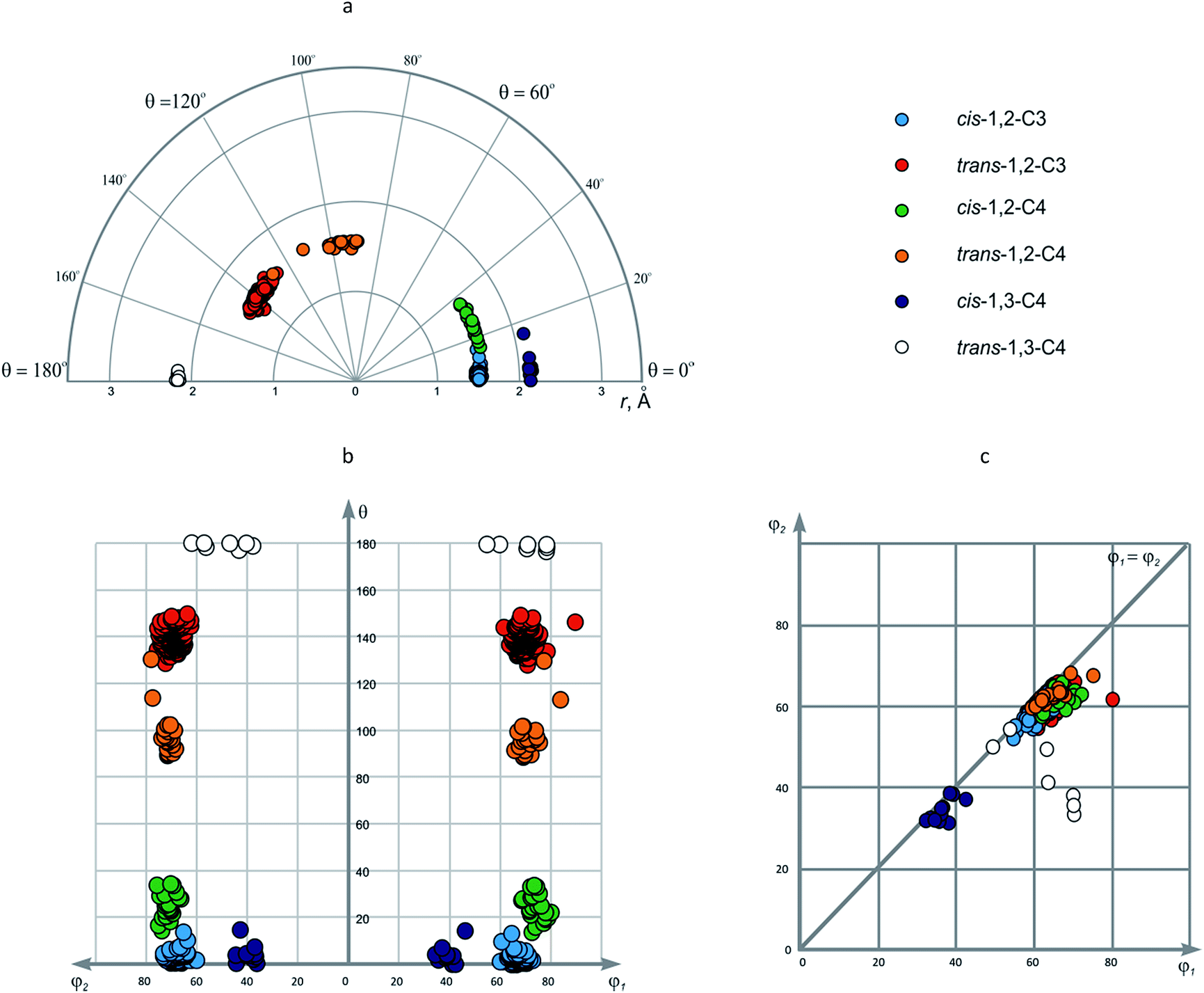 | ||
| Fig. 4 Disubstituted cyclopropanes and cyclobutanes shown in: (a) r–θ plot (polar coordinates); (b) θ–φ1/φ2 plot; (c) φ1–φ2 plot. | ||
| No. | Ring system | No. of points | r, Å | φ1, ° | φ2, ° | θ, ° |
|---|---|---|---|---|---|---|
| 1 | cis-1,2-C3 | 59 | 1.469–1.552 (1.513) | 58.7–65.3 (59.7) | 58.2–62.3 (57.6) | 0–13.2 (2.7) |
| 2 | trans-1,2-C3 | 246 | 1.362–1.604 (1.508) | 54.8–79.4 (61.5) | 53.4–64.9 (59.3) | 128.3–149.5 (139.9) |
| 3 | cis-1,2-C4 | 22 | 1.531–1.581 (1.557) | 61.3–71.6 (66.4) | 56.3–64.7 (60.8) | 13.8–33.8 (24.4) |
| 4 | trans-1,2-C4 | 22 | 1.476–1.591 (1.547) | 58.7–74.5 (63.5) | 58.4–67.0 (61.4) | 88.8–130.1 (97.7) |
| 5 | cis-1,3-C4 | 11 | 2.120–2.165 (2.139) | 31.7–41.9 (36.0) | 30.0–37.3 (33.0) | 0–14.3 (4.0) |
| 6 | trans-1,3-C4 | 7 | 2.141–2.189 (2.167) | 48.8–69.6 (62.3) | 32.0–53.1 (41.9) | 177.0–179.9 (179.1) |
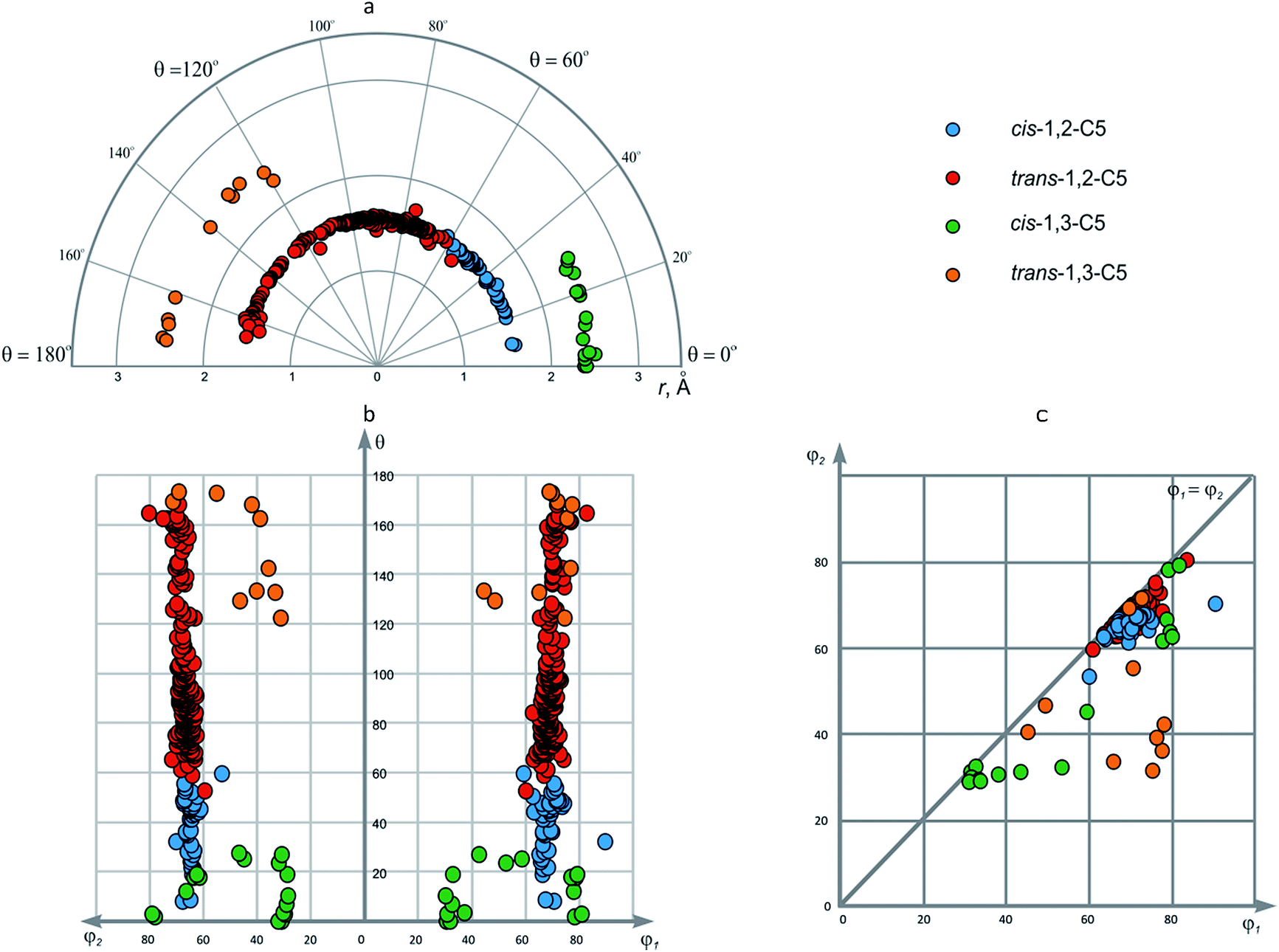
| 7 | cis-1,2-C5 | 44 | 1.475–1.617 (1.548) | 59.2–89.5 (68.8) | 52.8–69.8 (64.6) | 8.0–59.6 (39.8) |
| 8 | trans-1,2-C5 | 265 | 1.399–1.699 (1.539) | 60.0–82.6 (68.8) | 59.1–79.9 (66.8) | 52.7–168.5 (96.1) |
| 9 | cis-1,3-C5 | 21 | 2.375–2.503 (2.425) | 30.2–80.7 (56.4) | 28.3–78.7 (47.0) | 0–27.5 (12.9) |
| 10 | trans-1,3-C5 | 10 | 2.413–2.492 (2.450) | 44.4–77.2 (67.2) | 30.9–71.0 (46.0) | 122.6–173.6 (150.9) |
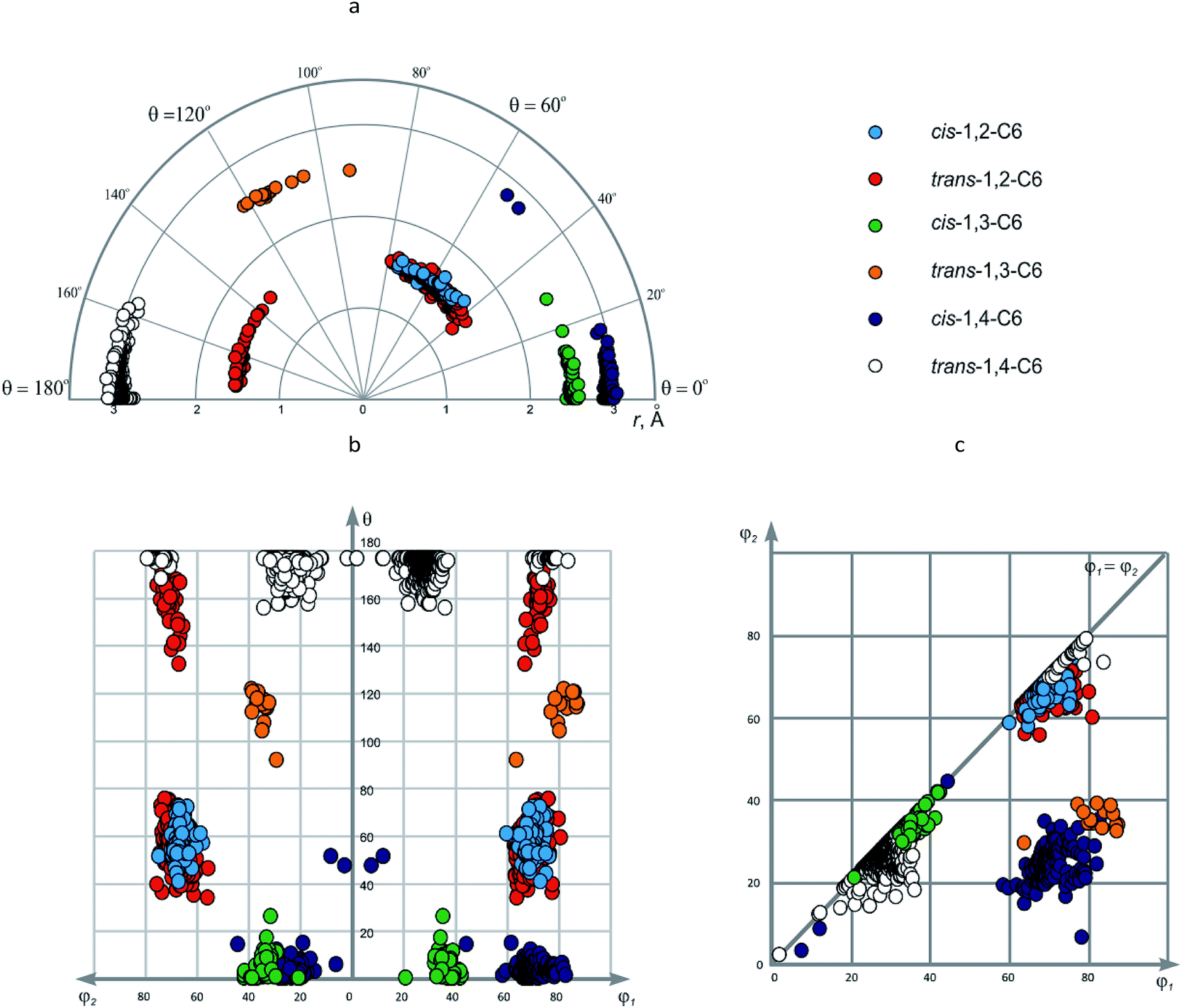
| 11 | cis-1,2-C6 | 174 | 1.434–1.667 (1.536) | 60.0–76.5 (69.3) | 57.9–74.5 (67.0) | 41.2–73.3 (57.4) |
| 12 | trans-1,2-C6 | 1117 | 1.328–1.651 (1.526) | 63.0–81.0 (70.4) | 55.8–75.7 (68.3) | 34.4–76.8; 134.6–174.8 (62.5) |
| 13 | cis-1,3-C6 | 57 | 2.451–2.600 (2.522) | 20.8–42.5 (36.0) | 20.8–41.8 (33.9) | 0–26.4 (5.0) |
| 14 | trans-1,3-C6 | 16 | 2.495–2.563 (2.532) | 63.7–87.5 (81.7) | 29.3–39.0 (35.0) | 93.4–123.8 (150.9) |
| 15 | cis-1,4-C6 | 197 | 2.815–3.057 (2.966) | 7.4–82.8 (69.8) | 2.9–44.4 (24.0) | 0–52.1 (3.9) |
| 16 | trans-1,4-C6 | 618 | 2.738–3.071 (2.960) | 1.9–83.8 (30.1) | 1.9–79.4 (27.9) | 158.7–180.0 (177.4) |
In φ1–φ2 plot, the data points form three areas, which correspond to 1,2-disubstituted cores (φ1/φ2 ∼ 60°), cis-1,3-C4 (φ1/φ2 ∼ 35°) and trans-1,3-C4 (φ1 ∼ 60°, φ2 ∼ 40°) (Fig. 4c). It is interesting to note that φ1–φ2 plot is very convenient to evaluate the dissymmetry of the scaffold since only φ1 and φ2 parameters can distinguish the variation points. In this view, symmetric cores are located at φ1 = φ2 line, whereas dissymmetric ones are drawn away from this position. In the case of small rings, the dissymmetry is observed for trans-1,3-C4 scaffold, which can be explained by different (i.e. pseudo-axial and pseudo-equatorial) orientation of the corresponding substituents in the puckered cyclobutane fragment (Fig. 5a, 1).
 | ||
| Fig. 5 Conformations of trans-1,3-disubstituted cyclobutanes according to CSD data. | ||
This is not true for two data points which refer to trans-1,3-cyclobutanedicarboxylic acid (2) and its disodium salt; in these cases, flat structure was reported by authors for the cyclobutane ring.57,58 Although revision of these data points from late 1960s might be necessary, it is important to note that the uncommon conformations were identified simply by visual inspection – an interesting aspect of potential application of EVP.
 | ||
| Fig. 6 Disubstituted cyclopentanes shown in: (a) r–θ plot (polar coordinates); (b) θ–φ1/φ2 plot; (c) φ1–φ2 plot. | ||
Due to the conformational flexibility of the cyclopentane ring mentioned above, it is not possible to outline some particular areas in φ1–φ2 plot. The data points for the 1,2-disubstituted cores correspond to the symmetric nature of the scaffolds, whereas for 1,3-disubstituted ones, both symmetric (3, 5, 7, 8) and non-symmetric (4, 6) representatives are encountered (Fig. 7). Again, simple visual inspection of EVP allowed for the classification of different cyclopentane conformations in this case.
 | ||
| Fig. 7 Conformations of (a) cis-; (b) trans-1,3-disubstituted cyclopentanes according to CSD data. | ||
 | ||
| Fig. 8 Disubstituted cyclohexanes shown in: (a) r–θ plot (polar coordinates); (b) θ–φ1/φ2 plot; (c) φ1–φ2 plot. | ||
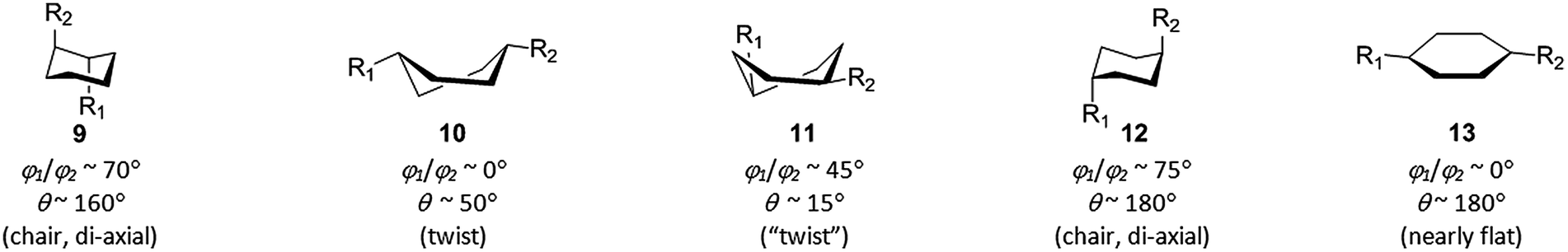 | ||
| Fig. 9 Uncommon conformations of disubstituted cyclohexanes revealed by EVP analysis of CSD data. | ||
Both cis- and trans-1,3-disubstituted cyclohexanes (cis- and trans-1,3-C6) do not show significant conformational heterogeneity, although it could be related to smaller number of the data points as compared to other representatives discussed in this section. The value of r is ∼2.5 Å, which again is slightly larger than in the case of 1,3-disubstituted derivatives of smaller rings. The values of other geometric parameters are: for cis-1,3-C6 – φ1/φ2 ∼ 35°, θ ∼0°; for trans-1,3-C6 – φ1 ∼ 80°, φ2 ∼ 35°, θ ∼ 120°; hence these isomers have very different conformational properties. These parameters describe favorable di-equatorial and axial-equatorial chair conformations, respectively. It is appropriate to note that the values of φ1/φ2 angles correlate well with axial or equatorial position of the corresponding substituent (∼80° for axial, ∼35° for equatorial). For both isomers, slightly deviating data points are observed, which correspond to the distorted conformations observed in metal complexes of cyclohexane-1,3-dicarboxylates.
As in the previous cases, most of the data points for 1,4-disubstituted cyclohexanes correspond to favorable axial-equatorial (cis-1,4-C6) and di-equatorial (trans-1,4-C6) chair conformations. The value of r is ∼2.95 Å – for obvious reason the largest among those discussed in this work. Other parameters for the above-mentioned chair conformers are: cis-1,4-C6 – φ1 ∼ 70°, φ2 ∼ 25°, θ ∼ 0°; trans-1,4-C6 – φ1/φ2 ∼ 25°; again, the values of φ1/φ2 angles correlate with axial (∼70°) and equatorial (∼25°) position of the corresponding substituents. The data points which do not fall into these common areas can be easily found by visual inspection of the EVP; this gives us twist conformations 10 and 11 for cis-1,4-C6, di-axial chair 12 (observed for 25 compounds) and flattened ring 13 (found in a nickel complex of cyclohexane-1,4-dicarboxylate) for trans-1,4-C6 (Fig. 9). In all these cases, it is steric strain which causes deformation of the cyclohexane ring and/or changes in position of the substituents.
According to φ1–φ2 plot, three areas are observed; two of them (φ1/φ2 ∼ 20–40° and ∼60–80°, respectively) correspond to symmetric scaffolds, namely, cis- and trans-1,2-C6, cis-1,3-C6 and trans-1,4-C6; the last one (φ1 ∼ 60–90°, φ2 ∼ 20–40°) – to non-symmetric, i.e. trans-1,3-C6 and cis-1,4-C6.
Combined EVP for the disubstituted cycloalkanes
In the previous sections, distribution of the data points in EVP was discussed considering the particular conformations of the corresponding cycloalkane scaffolds. Nevertheless, the main advantage of EVP is a possibility to give general characterization of conformational properties for large sets of diverse scaffolds. For more comprehensive analysis of the combined EVP for the disubstituted cycloalkanes, we have performed clustering of the data using a modification of the standard Jarvis–Patrick algorithm, which utilized variable-length nearest-neighbor lists.59,60 The clustering of the data was performed using Jarp tool of JChem package.61 To unify the coordinates for the metric distance function necessary for the clustering, the hyperspherical coordinate system defined by r, φ1, φ2, and θ was transformed to the Cartesian (x, y, z, t) (see our previous work46 for more details). Standard Euclidean distance d in this four-dimensional space was used to establish the dissimilarity of the data points. The maximum dissimilarity between the data points was found to be dmax = 6.122 Å. The threshold parameter T of the modified Jarvis–Patrick clustering was set to 10% of this value (0.612 Å), and the ratio of numbers of common neighbors for the two compounds Rmin – to 0.5. In addition, the T parameter was varied to be 5%, 3%, 2% and 1% of dmax in order to evaluate the stability of the clusters obtained.The results of the clustering are summarized in Table 2 and used for the coloring of the data points in Fig. 10. At the clustering parameter values given above, the data points split into seven clusters, three of which contain no more than two representatives. Four main clusters included 1949, 284, 630 and 18 compounds; we designated them as α, β, γ, and δ regions of the EVP, respectively. The cluster α is formed by all the 1,2-disubstituted cycloalkanes, and hence parameters r, φ1 and φ2 are not varied significantly within it: r ∼ 1.5 Å; φ1/φ2 ∼ 65°. On the contrary, for the θ angle, nearly all possible values within 0–180° range are observed. These results show that for 1,2-disubstituted cores, nearly all the possible mutual orientations of the substituents can be in principle achieved using monocyclic compounds; in this case, more complex analogs can be designed only to stabilize particular values of the θ angle.
| # | Cluster | Size | T, Å | r, Å | φ1, ° | φ2, ° | θ, ° |
|---|---|---|---|---|---|---|---|
| a Cartesian coordinates (x, y, z, t) in four-dimensional space, where x = rsinφ1sinφ2sinθ; y = rsinφ1cosφ2sinθ; z = rcosφ1sinθ; t = rcosθ. Maximum dissimilarity between the data points dmax = 6.122 Å. The threshold parameter value T = 1% or 10% of dmax (0.061 Å and 0.612 Å, respectively). The minimum ratio of numbers of common neighbors for the two compounds Rmin = 0.5. |
|||||||
| 1 | α | 1949 | 0.612 | 1.328–1.669 | 54.1–89.5 | 50.8–79.9 | 0–174.5 |
| 2 | α1A | 1411 | 0.061 | 1.435–1.593 | 63.0–76.4 | 60.3–73.2 | 41.2–103.7 |
| 3 | α1B | 57 | 0.061 | 1.469–1.552 | 54.1–65.3 | 50.8–62.3 | 0–6.7 |
| 4 | α1C | 24 | 0.061 | 1.542–1.576 | 64.0–71.6 | 59.1–65.6 | 16.0–30.9 |
| 5 | α2A | 248 | 0.061 | 1.447–1.570 | 56.7–73.6 | 53.4–70.0 | 131.2–148.4 |
| 6 | α2B | 48 | 0.061 | 1.439–1.601 | 67.5–76.8 | 65.1–74.6 | 149.5–164.4 |
| 7 | β | 284 | 0.612 | 2.120–3.057 | 20.8–82.8 | 6.1–78.7 | 0–27.5 |
| 8 | β1 | 188 | 0.061 | 2.898–3.057 | 58.4–82.8 | 14.4–35.2 | 0–9.2 |
| 9 | β2 | 58 | 0.061 | 2.377–2.600 | 20.8–42.5 | 20.8–41.8 | 0–12.0 |
| 10 | γ | 630 | 0.612 | 2.141–3.071 | 1.9–83.8 | 1.9–79.4 | 160.0–180.0 |
| 11 | γ | 596 | 0.061 | 2.738–3.071 | 1.9–83.8 | 1.9–79.4 | 168.4–180.0 |
| 12 | δ | 18 | 0.612 | 2.413–2.563 | 65.0–87.5 | 30.9–39.0 | 106.0–142.7 |
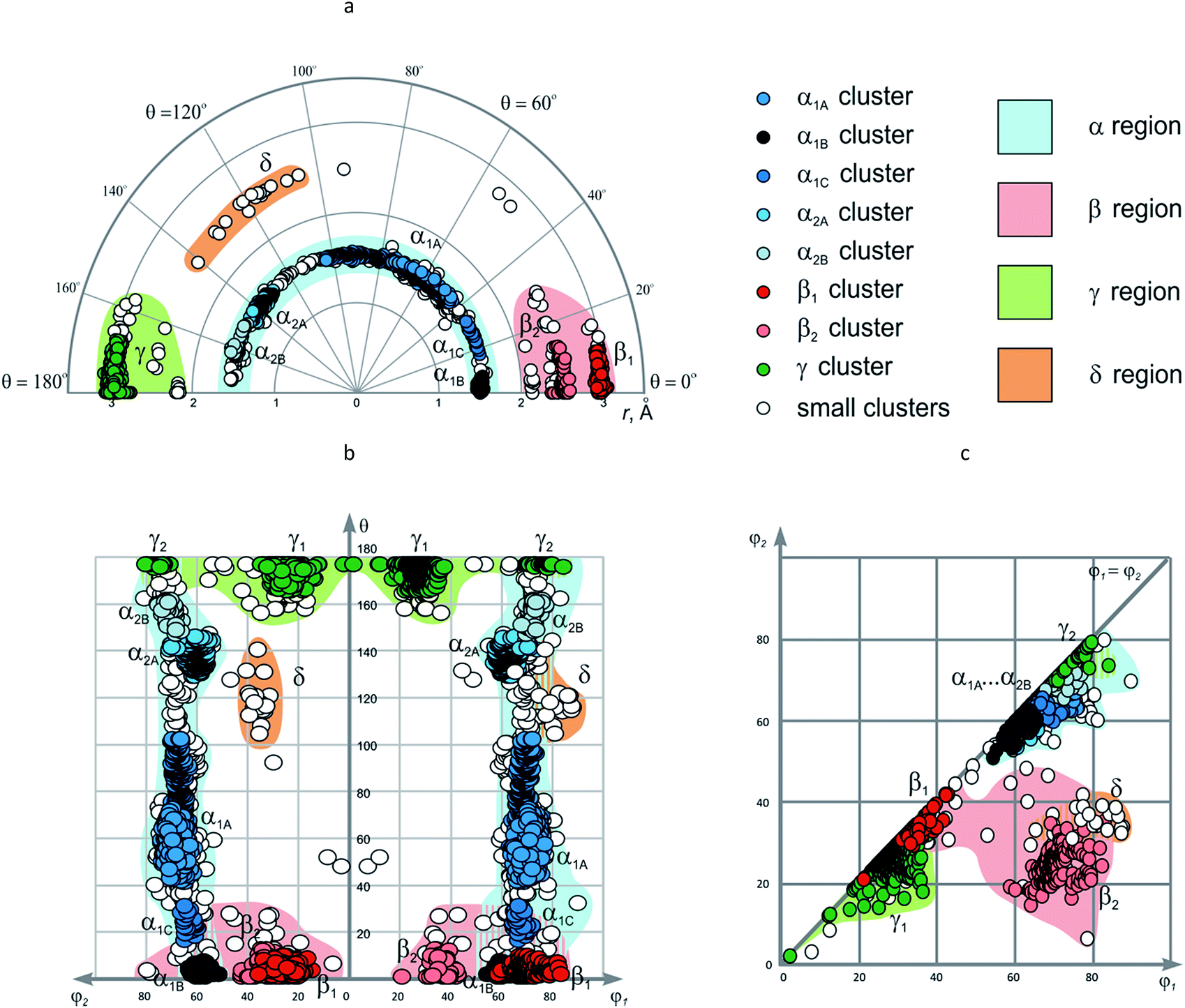 | ||
| Fig. 10 The combined EVP for disubstituted cycloalkanes: (a) r–θ plot (polar coordinates); (b) θ–φ1/φ2 plot; (c) φ1–φ2 plot. | ||
Under more strict clustering conditions (i.e. by decreasing T to 3% of dmax), the α cluster splits into two parts (Fig. 11) (some small clusters with no more than 10 members are also formed). Further decrease of T to 1% of dmax leads to additional cluster decomposition, so that five clusters with more than 10 compounds, as well as over 100 smaller ones are obtained (Table 2, Fig. 11). The largest of them, α1A, is formed by 1411 data points, which have θ value ∼40–100° and correspond to the following scaffolds: cis-1,2-C5, cis-1,2-C6, trans-1,2-C5, and trans-1,2-C6. Although the other clusters have smaller size, this might be related to lower occurrence of the corresponding cores in CSD, not to their lesser importance. Indeed, the clusters α1B (θ ∼ 0°), α1C (θ ∼ 20°), and α2A (θ ∼ 140°) correspond mainly to the cyclopropane and cyclobutane derivatives: cis-1,2-C3, cis-1,2-C4, and trans-1,2-C3, respectively, whereas α2B (θ ∼ 160°) is related to trans-1,2-C5 and trans-1,2-C6 cores with di-(pseudo)axial orientation of the substituents.
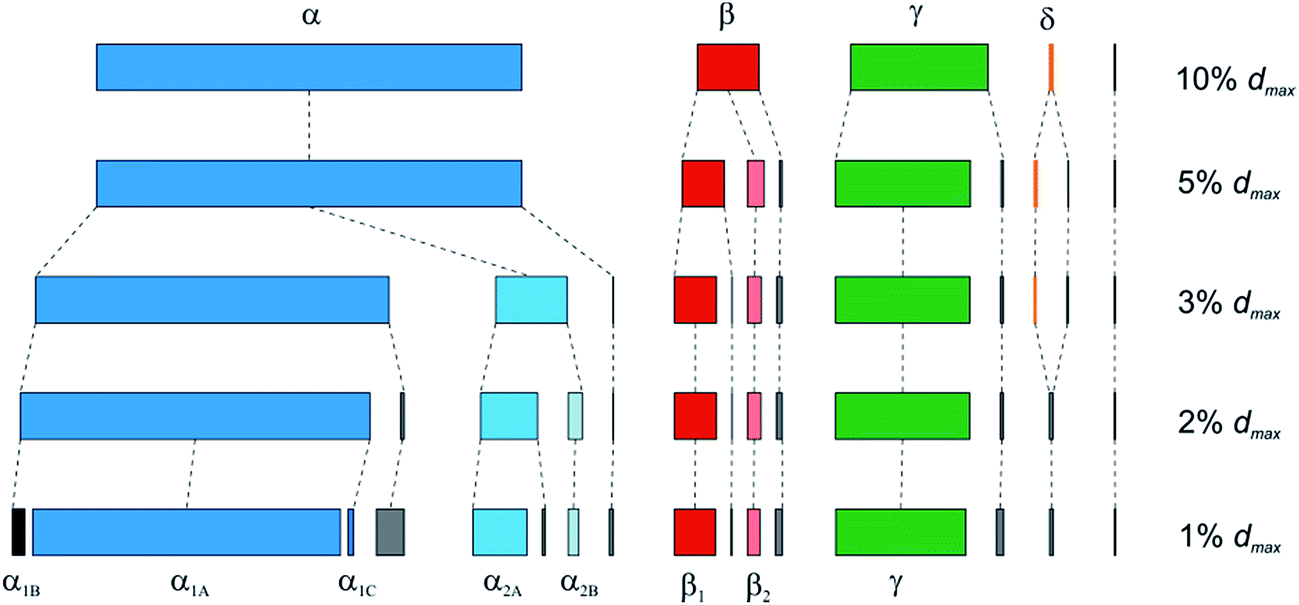 | ||
| Fig. 11 Clustering of the disubstituted cycloalkanes as a function of the threshold parameter T (varied from 10% to 1% of dmax). Rmin = 0.5, dmax = 6.122 Å. Small clusters with no more than 10 compounds are shown in grey. | ||
The β cluster is formed by cis-1,3-disubstituted scaffolds, as well as cis-1,4-C6. It shows less stability and splits into two parts β1 and β2 (and several small groups of data points) already at T = 5% of dmax. On the contrary, these subunits demonstrate significant stability towards the change of the clustering parameters up to T = 1% of dmax. The β1 (r ∼ 3.0 Å, φ1 ∼ 70°, φ2 ∼ 25°, θ ∼ 0°) and β2 (r ∼ 2.5 Å, φ1/φ2 ∼ 35°, θ ∼ 0°) clusters are formed by the data points derived almost exclusively from cis-1,4-C6 and cis-1,3-C6 scaffolds, respectively. Other cis-1,3-disubstituted scaffolds give only small clusters at this threshold parameter value.
Unlike its α and β counterparts, the γ cluster, which is formed by trans-1,4-C6 derivatives, demonstrates considerable stability towards the change of the clustering parameters: only a few small groups of data points split out upon decreasing T from 10% to 1% of dmax. Despite this stability, visual inspection of EVP shows that the compounds are distributed unevenly within the resulting γ cluster. Namely, two groups of the data points are observed which differ by φ1/φ2 values. The larger one (γ1, 573 rings) with φ1/φ2 ∼ 30° corresponds to the di-equatorial orientation of the substituents in the chair conformation of the cyclohexane, whereas the smaller one (γ2, 20 rings, φ1/φ2 ∼ 75°) – to the di-axial. This difference is clearly seen only in θ–φ1/φ2 and φ1–φ2 plots, since r and θ values do not show significant deviation from the average values (r ∼ 3.0 Å; θ ∼ 180°).
The δ cluster, which is formed only by 18 data points (at T = 10% of dmax), does not demonstrate sufficient stability towards more rigorous clustering conditions. Nevertheless, one of the possible reasons behind this could be the fact that it includes the derivatives of trans-1,3-C6 and (partially) trans-1,3-C5 cores, which are not numerous in CSD. That is why, in our opinion, the δ region of EVP, which corresponds to r ∼ 2.5 Å, φ1 ∼ 80°, φ2 ∼ 35°, θ ∼ 120°, is worth outlining.
Analysis of the β, γ, and δ regions of EVP shows that unlike the α area, they do not cover all the values of θ angle. In particular, the θ ∼ 40–100° range, which is represented well in the α region of the map, is virtually empty for larger r values characteristic for the β, γ, and δ areas. Therefore, design of the scaffolds with r > 2 Å can be aimed at filling such empty regions.
In the φ1–φ2 plot, the clusters discussed above are not observed separately. In particular, all subunits of the α region overlap with each other, as well as the γ2 group and many small clusters with φ1/φ2 ∼ 60–80°. Analogously, β1 and γ1 clusters (φ1/φ2 ∼ 20–40°) are also overlapped. All these data sets correspond to the symmetric scaffolds; in the case of non-symmetric ones, the two areas, namely, β2 and δ, can in principle be distinquished (φ1 ∼ 60–85°, φ2 ∼ 20–40°). It is worth noting that such non-symmetric disubstituted cycloalkane scaffolds are not numerous in CSD.
Conclusions and perspectives
Exit vector plot (EVP) analysis can be a useful tool for study and visualization of conformational properties of three-dimensional scaffolds. It is based on very simple geometrical model and can rely on either theoretical or experimental data on atomic coordinates of the molecules. EVP analysis of the CSD data for disubstituted cycloalkanes revealed four clearly defined and isolated regions in the diagrams (α, β, γ, and δ areas), unlike in the case of bicyclic conformationally restricted diamines studied in our previous work.46 The EVP analysis can be used in various areas where spatial arrangement of molecular fragments is important, especially those where intermolecular interactions are involved, including medicinal, supramolecular and coordination chemistry, asymmetric synthesis and conformational analysis. Possible directions of EVP use are listed below:• EVP analysis gives an opportunity for easy identification of unusual conformers with abnormal geometric parameters. Fig. 9 of this work illustrates some examples.
• EVP analysis of disubstituted cycloalkanes (as well as other simple ring systems) can be used as a guide for the design of more complex (e.g. bicyclic) scaffolds. In particular, we have shown that 1,2-disubstituted cores (α area of EVP) evenly cover the theoretically accessible conformational space, hence their analogues can be used only to stabilize particular conformations. For other disubstituted cycloalkanes with r > 2 Å, many empty regions are found, and the respective bi- or polycyclic analogues might fill these gaps.
• EVP can be used for classification of conformational space covered by disubstituted scaffolds, regardless of their composition. This could be especially useful for the analysis of large data sets related to biological activity (e.g. compound libraries in medicinal chemistry). In our opinion, the α, β, γ, and δ regions defined in this work might be extended by further data, but their principal location in the diagram should not change significantly. In particular, the data points for the cis- and trans-1,2-disubstituted cycloheptanes available from CSD fit well into the α area, analogously to the corresponding cis- and trans-1,2-C6 derivatives (Fig. 12). For the trans-1,4-C7 scaffold, two data points are available: one can be considered as analogous to γ area, another does not fall into any of the regions introduced. Possibly, it might fall into extended δ area which included larger r values.
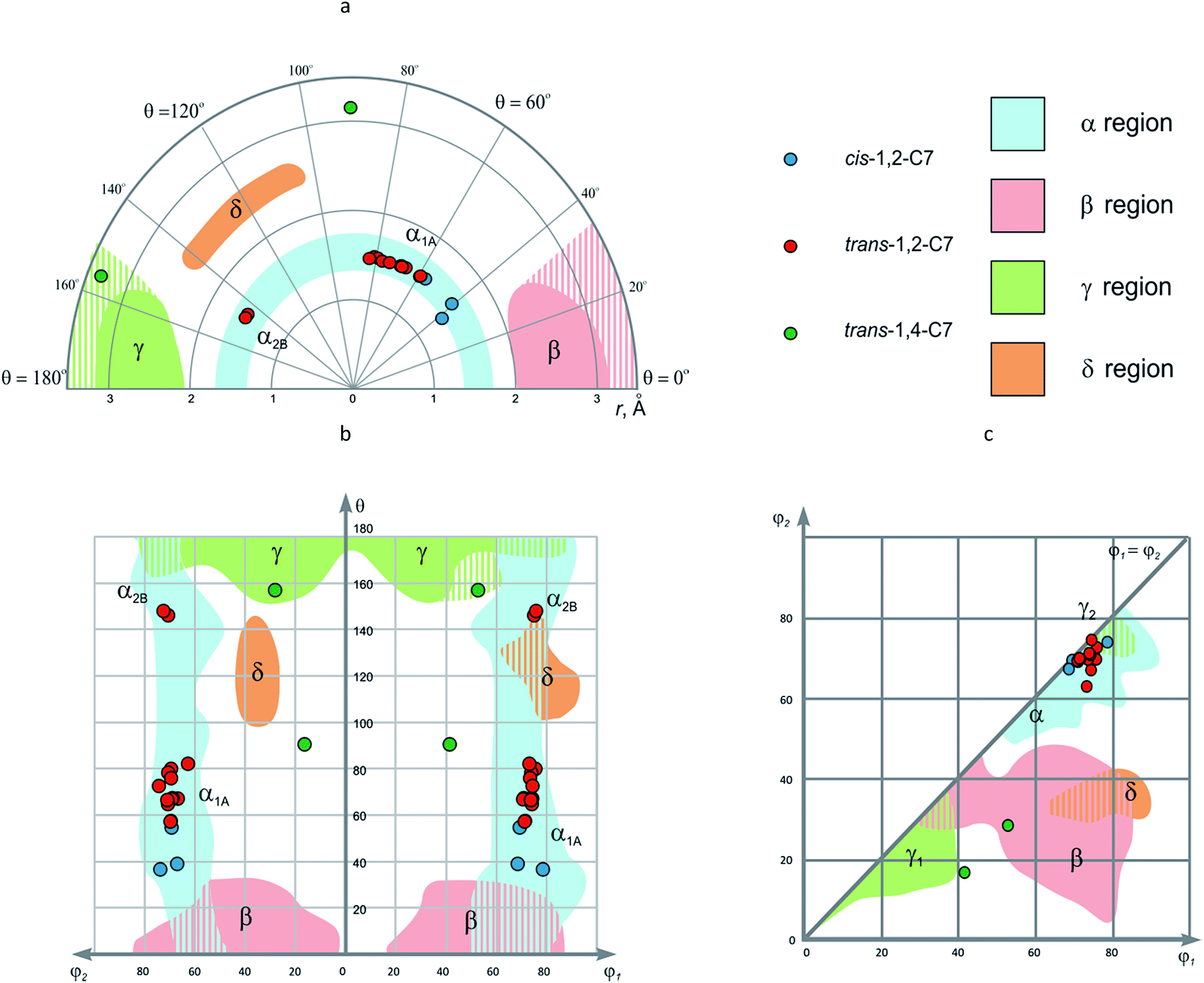 | ||
| Fig. 12 The EVP for disubstituted cycloheptanes: (a) r–θ plot (polar coordinates); (b) θ–φ1/φ2 plot; (c) φ1–φ2 plot. | ||
• Rational scaffold replacement is possible with the guide of EVP data. In particular, we have recently shown that various isomers of 1,6-disubstituted spiro[3.3]heptanes can be potential surrogates of either cis-1,4-C6 or trans-1,3-C6 scaffolds.48 Using the terms introduced in this work, the corresponding isomers fall into β and δ areas of the EVP.
• EVP data can assist in building structure–property relationships, if spatial arrangement of the functional groups is the factor which affects significantly the particular property. This aspect is important first of all to medicinal chemistry, but it can be also used for prediction of e.g. physico-chemical parameters. Recently, we have shown that Euclidean distance d in (x, y, z, t) space (easily obtained from r, φ1, φ2, θ coordinates) shows good correlation with biological activity for nicotine acetylcholine receptor ligands.46 Notably, for the fluoroquinolone antibiotics, where arrangement of the substituent around the diamine scaffold does not affect biological activity significantly since it is not involved into the mechanism of action, no such correlation was observed. In this work, we give one more example derived from cycloalkane scaffolds discussed in this work, namely, chemokine receptor CKCR3 antagonists 14 (Fig. 13) taken from the Sanofi patent.62 The parameters r, φ1, φ2, θ for these compounds were evaluated using average values for the corresponding scaffolds given in Table 1. A correlation with r2 = 0.515 was found between relative biological activity and parameter d for these series, which shows that relative orientation of the functional groups attached to the cycloalkane scaffold plays important role in interaction of 14 with their biological target. For this particular case, the optimal cycloalkane scaffolds are located near γ region of EVP.63
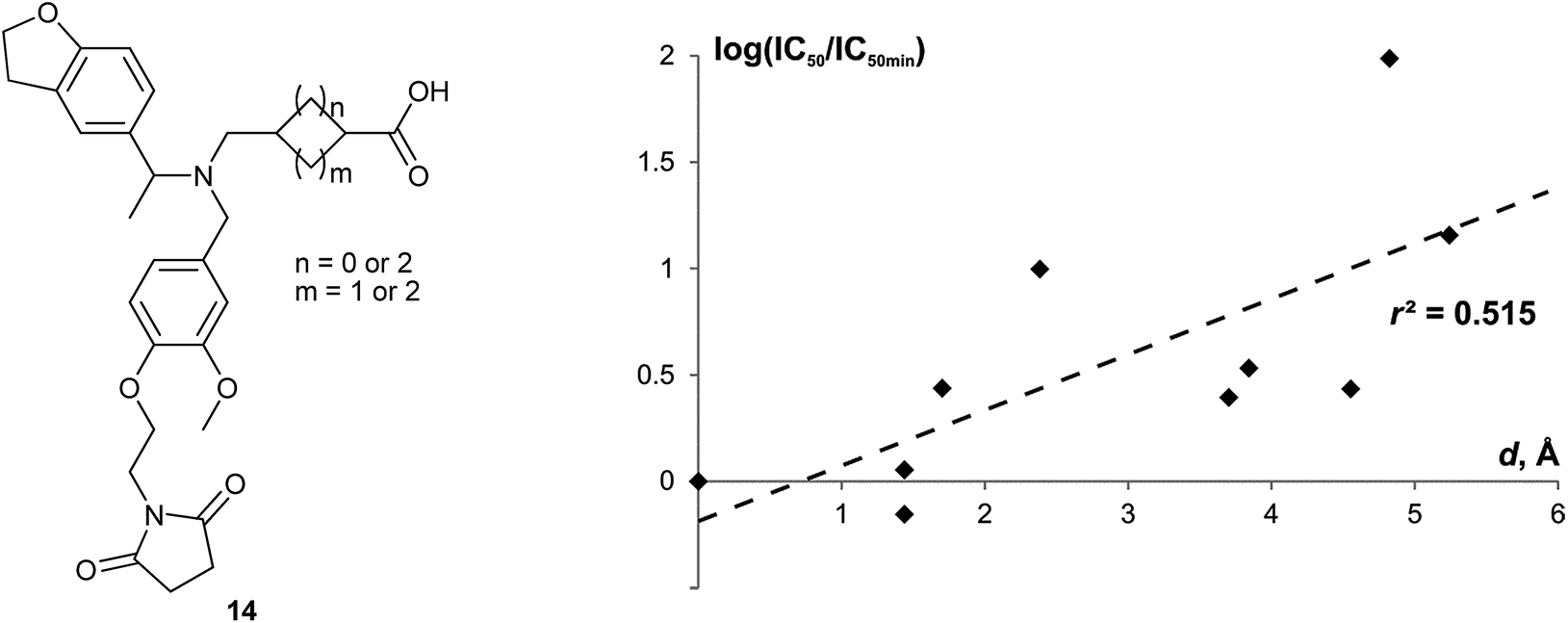 | ||
| Fig. 13 Correlation between the relative biological effect of the chemokine receptor CXCR3 antagonists 14 (log(IC50/IC50min)) and the Euclidean distance of the corresponding cycloalkane scaffolds from the scaffold of the most active compound (d).64 | ||
To help the readers in using exit vector plots (EVP) for their own analysis of scaffolds, we have provided a program which calculates all the necessary geometric parameters (r, φ1, φ2, θ) from SDF containing 3D coordinates (see ESI for more details).
Notes and references
- P. Kirkpartick and C. Ellis, Nature, 2004, 432, 823 CrossRef.
- C. M. Dobson, Nature, 2004, 432, 824 CrossRef CAS PubMed.
- J.-L. Reymond, R. van Deursen, L. C. Blum and L. Ruddigkeit, Med. Chem. Commun., 2010, 1, 30 RSC.
- D. J. Triggle, Biochem. Pharmacol., 2009, 78, 217 CrossRef CAS PubMed.
- L. Eberhardt, K. Kumar and H. Waldmann, Curr. Drug Targets, 2011, 12, 1531 CrossRef CAS PubMed.
- J. L. Medina-Franco, Chemoinformatic characterization of the chemical space and molecular diversity of compound libraries, in Diversity-oriented synthesis: basics and applications in organic synthesis, drug discovery, and chemical biology, ed A. Trabocchi, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2013 Search PubMed.
- J. L. Medina-Franco, Drug Dev. Res., 2012, 73, 430 CrossRef CAS.
- Y.-S. Wong, Methods Mol. Biol., 2012, 800, 11 CAS.
- M. Dow, M. Fisher, T. James, F. Marchetti and A. Nelson, Org. Biomol. Chem., 2012, 10, 17 CAS.
- M. A. Koch, A. Schuffenhauer, M. Scheck, S. Wetzel, M. Casaulta, A. Odermatt, P. Ertl and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17272 CrossRef CAS PubMed.
- J. L. Medina-Franco, K. Martinez-Mayorga, M. A. Giulianotti, R. A. Houghten and C. Pinilla, Curr. Comput.–Aided Drug Des., 2008, 4, 322 CrossRef CAS.
- R. van Deursen, L. C. Blum and J.-L. Reymond, J. Comput.–Aided Mol. Des., 2011, 25, 649 CrossRef PubMed.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887 CrossRef CAS PubMed.
- S. R. Langdon, N. Brown and J. Blagg, J. Chem. Inf. Model., 2011, 51, 2174 CrossRef CAS PubMed.
- M. Krier, G. Bret and D. Rognan, J. Chem. Inf. Model., 2006, 46, 512 CrossRef CAS PubMed.
- P. Ertl, A. Schuffenhauer and S. Renner, Methods Mol. Biol., 2011, 672, 245 CAS.
- R. D. Taylor, M. MacCoss and A. D. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed.
- V. Khanna and S. Ranganathan, J. Cheminf., 2011, 3, 30 CAS.
- D. K. Agrafiotis and J. J. Wiener, J. Med. Chem., 2010, 53, 5002 CrossRef CAS PubMed.
- H. Zhao and I. Akritopoulou-Zanze, Expert Opin. Drug Discovery, 2010, 5, 123 CrossRef CAS PubMed.
- S. Wetzel, K. Klein, S. Renner, D. Rauh, T. I. Oprea, P. Mutzel and H. Waldmann, Nat. Chem. Biol., 2009, 5, 581 CrossRef CAS PubMed.
- S. N. Pollock, E. A. Coutsias, M. J. Wester and T. I. Oprea, J. Chem. Inf. Model., 2008, 48, 1304 CrossRef CAS PubMed.
- M. J. Wester, S. N. Pollock, E. A. Coutsias, T. K. Allu, S. Muresan and T. I. Oprea, J. Chem. Inf. Model., 2008, 48, 1311 CrossRef CAS PubMed.
- C. M. Marson, Chem. Soc. Rev., 2011, 40, 5514 RSC.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed.
- K. Kingwell, Nat. Rev. Drug Discovery, 2009, 8, 931 CrossRef CAS PubMed.
- A. Nicholls, G. B. McGaughey, R. P. Sheridan, A. C. Good, G. Warren, M. Mathieu, S. W. Muchmore, S. P. Brown, J. A. Grant, J. A. Haigh, N. Nevins, A. N. Jain and B. Kelley, J. Med. Chem., 2010, 53, 3862 CrossRef CAS PubMed.
- M. Aldeghi, S. Malhotra, D. L. Selwood and A. W. Chan, Chem. Biol. Drug Des., 2014, 83, 450 CAS.
- F. Lovering, Med. Chem. Commun., 2013, 4, 515 RSC.
- W. H. Sauer and M. K. Schwarz, J. Chem. Inf. Comput. Sci., 2003, 43, 987 CrossRef CAS PubMed.
- N. C. Firth, N. Brown and J. Blagg, J. Chem. Inf. Model., 2012, 52, 2516 CrossRef CAS PubMed.
- J. A. Wilson, A. Bender, T. Kaya and P. A. Clemons, J. Chem. Inf. Model., 2009, 49, 2231 CrossRef CAS PubMed.
- P. J. Ballester and W. G. Richards, J. Comput. Chem., 2007, 28, 1711 CrossRef CAS PubMed.
- V. Venkatraman, P. R. Chakravarthy and D. Kihara, J. Cheminf., 2009, 1, 19 Search PubMed.
- A. S. Karaboga, F. Petronin, G. Marchetti, M. Souchet and B. Maigret, J. Mol. Graphics Modell., 2013, 41, 20 CrossRef CAS PubMed.
- C. M. Richardson, M. J. Lipkin and D. W. Sheppard, Bioorg. Med. Chem. Lett., 2015, 25, 2089 CrossRef CAS PubMed.
- T. Schulz-Gasch, C. Schärfer, W. Guba and M. Rarey, J. Chem. Inf. Model., 2012, 52, 1499 CrossRef CAS PubMed.
- D. C. Kombo, K. Tallapragada, R. Jain, J. Chewning, A. A. Mazurov, J. D. Speake, J. D. Hauser and S. Toler, J. Chem. Inf. Model., 2013, 53, 327 CrossRef CAS PubMed.
- S. C. Lovell, I. W. Davis, W. B. Arendall, P. I. W. De Bakker, J. M. Word and M. G. Prisant, Proteins: Struct., Funct., Genet., 2003, 50, 437 CrossRef CAS PubMed.
- G. N. Ramachandran, C. Ramakrishnan and V. Sasisekharan, J. Mol. Biol., 1963, 7, 95 CrossRef CAS PubMed.
- A. Banerjee and P. Balaram, Curr. Sci., 1997, 73, 1067 CAS.
- S. J. Cottrell, T. S. G. Olsson, R. Taylor, J. C. Cole and J. W. Liebeschuetz, J. Chem. Inf. Model., 2012, 52, 956 CrossRef CAS PubMed.
- R. Taylor, J. Cole, O. Korb and P. McCabe, J. Chem. Inf. Model., 2014, 54, 2500 CrossRef CAS PubMed.
- C. Schärfer, T. Schulz-Gasch, H.-C. Ehrlich, W. Guba, M. Rarey and M. Stahl, J. Med. Chem., 2013, 56, 2016 CrossRef PubMed.
- G. Lauri and P. A. Bartlett, J. Comput.–Aided Mol. Des., 1994, 8, 51 CrossRef CAS PubMed.
- O. O. Grygorenko, R. Prytulyak, D. M. Volochnyuk, V. Kudrya, O. V. Khavryuchenko and I. V. Komarov, Mol. Diversity, 2012, 16, 477 CrossRef CAS PubMed.
- D. S. Radchenko, S. O. Pavlenko, O. O. Grygorenko, D. M. Volochnyuk, S. V. Shishkina, O. V. Shishkin and I. V. Komarov, J. Org. Chem., 2010, 75, 5941 CrossRef CAS PubMed.
- A. V. Chernykh, D. S. Radchenko, O. O. Grygorenko, C. G. Daniliuc, D. M. Volochnyuk and I. V. Komarov, J. Org. Chem., 2015, 80, 3974 CrossRef CAS PubMed.
- V. S. Yarmolchuk, I. L. Mukan, O. O. Grygorenko, A. A. Tolmachev, S. V. Shishkina, O. V. Shishkin and I. V. Komarov, J. Org. Chem., 2011, 76, 7010 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2002, 58, 380 CrossRef.
- F. H. Allen, O. Kennard and R. Taylor, Acc. Chem. Res., 1983, 16, 146 CrossRef CAS.
- H. L. Strauss and H. M. Pickett, J. Chem. Phys., 1971, 55, 324 CrossRef.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354 CrossRef CAS.
- N. S. Zefirov, V. A. Palyulin and E. E. Dashevskaya, J. Phys. Org. Chem., 1990, 3, 147 CrossRef CAS.
- P. Khalili, C. B. Barnett and K. J. Naidoo, J. Chem. Phys., 2013, 138, 184110 CrossRef PubMed.
- N. M. O′Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 4, 33 Search PubMed.
- T. N. Margulis and M. S. Fisher, J. Am. Chem. Soc., 1967, 89, 223 CrossRef CAS.
- E. Adman and T. N. Margulis, J. Am. Chem. Soc., 1968, 90, 4517 CrossRef CAS.
- R. A. Jarvis and E. A. Patrick, IEEE Trans. Comput., 1973, C22, 1025 CrossRef.
- J. M. Barnard and G. M. Downs, J. Chem. Inf. Comput. Sci., 1997, 37, 141 CrossRef CAS.
- JChem, version 15.3.16.0, build date, 2015-03-16, ChemAxon, Budapest, Hungary, http://www.chemaxon.com, 2015 Search PubMed.
- I. Bata, P. Buzder-Lantos, A. Vasas, B. V. Bartané, G. Ferenczy, Z. Tömösközi, G. Szeleczky and S. Batori. Eur. Pat. 2601950, 2013.
- To be more precise, it is (−)-γ region, where (−) is for the sign of θ value which should be taken into account for biological activity since it describes chirality of the molecule.
- The relative biological effect is measured as log(IC50/IC50min), where IC50min is the IC50 value for the most active compound. The Euclidean distance d is determined in the Cartesian space (x, y, z, t); x = rsinφ1sinφ2sinθ; y = rsinφ1cosφ2sinθ; z = rcosφ1sinθ; t = rcosθ. The sign of θ angle was taken into account for different enantiomers. For the mixtures of stereoisomers, average values of d were used.
Footnote |
| † Electronic supplementary information (ESI) available: The program which allows calculation of EVP geometric parameters from SDF containing 3D coordinates, together with sample files. See DOI: 10.1039/c5ra19958a |
| This journal is © The Royal Society of Chemistry 2016 |