
Mesoporous transition metal dichalcogenide ME2 (M = Mo, W; E = S, Se) with 2-D layered crystallinity as anode materials for lithium ion batteries†
Abstract
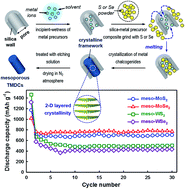
Mesoporous transition metal dichalcogenides (TMDCs), composed of group VI metals (Mo and W) and chalcogens (S and Se), with 2-D layered crystalline frameworks and 3-D pore structures were successfully prepared via a melting-infiltration assisted nano-replication method using a mesoporous template KIT-6 with cubic Ia3d symmetry. Combined analysis using X-ray diffraction, N2 adsorption–desorption and electron microscopy indicated that the mesoporous TMDCs, thus obtained, exhibited high surface areas (87–105 m2 g−1), large pore volumes (0.21–0.25 cm3 g−1) and well-defined mesopores about 20 nm in diameters. The mesoporous TMDCs showed outstanding rate capabilities up to 2C as well as high reversible lithium storage capacities (MoS2 710 mA h g−1; MoSe2 744 mA h g−1; WS2 501 mA h g−1; WSe2 427 mA h g−1) without a remarkable fading of capacity.
 Please wait while we load your content...
Please wait while we load your content...

