
Biodegradable polyurethane acrylate/HEMA-grafted nanodiamond composites with bone regenerative potential applications: structure, mechanical properties and biocompatibility
Abstract
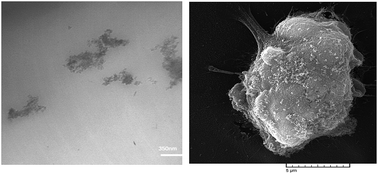
The present study demonstrates HEMA-grafted nanodiamond (ND-HEMA)/acrylate-terminated polyurethane-acrylate diluents (APUA) composites as promising materials for bone implant applications. Neat APUA and APUA composites containing ND-HEMA at different loadings up to 2 wt% were prepared by an in situ polymerization method. Morphological analysis demonstrated that ND-HEMAs were actually in the form of tightly bound aggregates which led to formation of big agglomerates at a concentration of 2 wt%. It was also suggested that ND-HEMAs were preferentially localized in the continuous soft domain of APUA; however it interacted by both soft and hard domains. Moreover, ND-HEMAs caused considerable phase separation between soft and hard domains as well as increased crystallinity. Maximum improvement in tensile properties of APUA was observed at 1 wt% loading of ND-HEAMs, namely 175% improvement in modulus and 40% increase in strength. The hydrophilic nature of ND-HEMA enhanced water absorption of composites resulted in higher hydrolysis degradation of APUA. In vitro biocompatibility evaluation via culturing human osteosarcoma cells (MG-63 osteoblast-like cell line), demonstrated no adverse effect on the cell viability of samples. Furthermore, the composites showed favorable cell adhesion and growth, and ND-HEMAs did not cause any negative effect on proliferation, ALP production and osteoblast attachment by MG-63 cells compared with neat APUA.
 Please wait while we load your content...
Please wait while we load your content...

