
Influence of plasma macronutrient levels on hepatic metabolism: role of regulatory networks in homeostasis and disease states†
Abstract
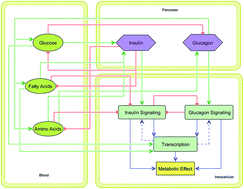
The human liver acts as a homeostatic controller for maintaining the normal levels of plasma metabolite concentrations by uptake, utilization, storage and synthesis of essential metabolites. These hepatic functions are orchestrated through a multilevel regulation composed of metabolic, signaling and transcriptional networks. Plasma macronutrients namely, glucose, amino acids and fatty acids are known to influence these regulatory mechanisms to facilitate homeostasis. We composed a regulatory circuit that elicits the design principle behind the metabolic regulation in liver. We have developed a detailed dynamic model for hepatic metabolism incorporating the regulatory mechanisms at signaling and transcriptional level. The model was analyzed to capture the behavior of hepatic metabolic fluxes under various combinations of plasma macronutrient levels. The model was used to rationalize and explain the experimental observations of metabolic dysfunctions through regulatory mechanisms. We addressed the key questions such as, how high carbohydrate diet increases cholesterol and why a high protein diet would reduce it; how high fat and high protein diet increases gluconeogenesis leading to hyperglycemia; how TCA (tricarboxylic acid) cycle is impaired through diet induced insulin resistance; how high fat can impair plasma ammonia balance; how high plasma glucose can lead to dyslipidemia and fatty liver disease etc. The analysis indicates that higher levels (above 2.5–3 fold) of macronutrient in plasma results in impairment of metabolic functions due to perturbations in the regulatory circuit. While higher glucose levels saturate the rate of plasma glucose uptake, higher amino acids activate glucagon and inhibit IRS (insulin receptor substrate) through S6K (S6 kinase), whereas higher fatty acid levels inhibit IRS through DAG–PKC (diacylglycerol and protein kinase C) and TRB3 activation. Moreover the ATP–ADP ratio is reduced under such conditions and β-oxidation is up-regulated through activation of PPARα (peroxisome proliferator-activated receptor alpha) leading to reduced anabolic capacity and increased cataplerosis in TCA cycle. The above factors together decrease insulin sensitivity and enhances glucagon effect through underlying signaling and transcriptional network leading to insulin resistance in liver. Such a metabolic state is known to result in diabetes and non-alcoholic fatty liver disease.
 Please wait while we load your content...
Please wait while we load your content...

