
Direct transition mechanism for molecular diffusion in gas hydrates
Abstract
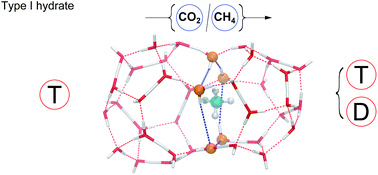
Several mechanisms for transport of gases inside hydrates have been proposed in the literature. Previous results have pointed out the apparent impossibility of some guests passing directly through faces connecting adjacent cages without destroying the water structure. Alternative diffusion mechanisms were proposed, such the participation of an additional guest (help-gas) or a water-hopping displacement of defects in the lattice. In this work, we use dual cage explicit atomic systems, where the position of some external atoms are fixed, to demonstrate theoretically that direct transitions are feasible through hexagonal and pentagonal faces in type I hydrate, for both carbon dioxide and methane, without compromising the overall structure integrity. Our DFT calculations show that even in the case that some bonds break during the transition, all of them are recovered, because the face distortion is absorbed locally by the hydrogen bond network. This result opens the door to improve the description of transport mechanisms of guest molecules by means of direct inter-cage transitions.
 Please wait while we load your content...
Please wait while we load your content...

