
New insight into the difference of PC lipase-catalyzed degradation on poly(butylene succinate)-based copolymers from molecular levels
Jia-xiang Qin*a,
Min Zhanga,
Chi Zhanga,
Cheng-tao Lia,
Yi Zhanga,
Jie Songa,
Hafiz M. Asif Javedb and
Jian-hui Qiuc
aKey Laboratory of Auxiliary Chemistry & Technology for Chemical Industry, Ministry of Education, Shaanxi University of Science & Technology, Xi'an 710021, China. E-mail: qinwushang@163.com
bElectronic Materials Research Laboratory, International Center for Dielectric Research, Xi'an Jiaotong University, Xi'an 710049, China
cAkita Prefecture University, Akita 015-0055, Japan
First published on 29th January 2016
Abstract
The homopolymer poly(butylene succinate) (PBS) and its copolymers containing 1,4-cyclohexane dimethylene succinate (CHDMS) or butylene 1,4-cyclohexanedicarboxylate (BCHDA) sequences with different molecular architectures (P(BS-co-CHDMS) and P(BS-co-BCHDA)) were prepared via melt polycondensation in the presence of a Ti-based catalyst. With the aim to gain more underlying information about enzymatic degradation rules and differences, the enzymatic degradation studies were performed in chloroform using Pseudomonas cepacia lipase (PCL) for 60 h. Degradation was monitored using various analytical techniques such as GPC, TGA and MALDI-TOF-MS. The results show that all PBS-based copolymers had an obvious degradation in chloroform. Compared with P(BS-co-BCHDA), P(BS-co-CHDMS) showed greater degradation rates after 60 hours of enzymatic degradation. The maximum degradation percentage observed in P(BS-co-10%CHDMS) was about 85%. Similarly, thermal property changes were observed with a decrease of the decomposition temperature of 5% and 15% sample in most cases. The enzymatic degradation of PBS-based copolymers produced not only linear segments, but also cyclic oligomers. Furthermore, P(BS-co-CHDMS) produced more oligomers than PBS-co-BCHDA. According to the results of molecular docking, the free energy of binding between PCL and the substrate in chloroform was in the order CMSCM > BSCM > BCAB > BSB. That is, the docking of the substrate containing CHDMS in the active site of PCL was more stable than any other ones.
Introduction
Polyesters have been extensively applied in recent years for biomaterials, packaging materials, agriculture, gene delivery, drug delivery, and tissue engineering,1–7 due to their biodegradability and biocompatibility. Despite such advantages, the overuse of polyesters will lead to the waste of carbon resources beyond redemption. In addition, critical shortages of fossil oil and other natural resources will occur in the near future, and the potential demands for recycling of polymeric materials and recovery of value-added carbon resources have increased worldwide in both social and environmental viewpoints. However, chemical recycling of polymeric materials is limited because of their prohibitively higher energy consumption and longer downstream processing. Therefore it is urgent to the development of an ecofriendly and speedy way for recycling of polymeric materials.8 According to Shiro Kobayash,9 enzymatic degradation of the polymer in organic solvents is a new effective method for carbon resource recycling. There are many advantages in the degradation of polymeric materials with enzyme in organic solvent, such as simple operation, less influential factors and convenient product separation. It is of great significance and potential industrial application to regard the enzymatic degradation product as a resource recycling for reuse.9–11Lipase is one of the most important enzymes because of its high stability and strong activity in nearly water-immiscible organic solvents. As a result, lipase has evolved to be efficient catalysts for lipolytic reactions involving in the hydrolysis of ester linkages.12,13 Several seminal works have been reported for the application of Lipases in the degradation of polyesters8,14–17 and polyurethanes18 in organic solvents. On the contrary, little work has been done on the perceptibility of Pseudomonas cepacia lipase on ester bond of different copolyesters in organic solvent with molecular docking and molecular dynamics simulations and insight the degradation mechanism at the atomic level.
Compared with traditional experimental methods, the molecular dynamic simulation can reproduce structural changes in the process of protein at the scale of nanoseconds with the help of powerful computing capabilities of computer, and provide detailed information about changes of the protein structure at the atomic level, which can be used to assist and guide traditional experiments.19–24 The present work aimed to study degradation of polyesters catalyzed by lipase from Pseudomonas cepacia in chloroform at molecular level. Combining docking and molecular dynamics simulations, we investigated the binding modes of a model polyester molecule within PCL. Positions and interactions of substrates with the active site of PCL were examined and the proximity of ester bond to the catalytic residues was analyzed. As a comparison, two polyester molecules were chosen as model substrates for catalyze degradation in this work. This work provided the first step for the insight of enzymatic degradation mechanism of copolyesters in the chloroform. If achieved, this goal would be a substantial step forward for rapid recycling of carbon resource.
Materials and methods
Materials
PBS, P(BS-co-CHDMS), and P(BS-co-BCHDA) were synthesized as previous report.25 Chloroform (CHCl3) was analytically pure, purchased from Xi'an Chemical Reagent Co. Ltd. 1,4-Succinic acid (SA), 1,4-butanediol (BDO), 1,4-cyclohexane dimethanol (CHDM), 1,4-cyclohexanedi-carboxylic acid (CHDA) and other reagents were analytically pure and purchased from Alfa Aesar Chemical Reagent Co. Ltd. Immobilized lipase from Pseudomonas cepacia (PCL) 900 U g−1, was purchased from Sigma-Aldrich Co. LLC. All reagents were directly used without further purification.Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BDO (mol:mol = 1:1.1) were added into the three-necked flask with nitrogen flow, then Ti(OiPr)4 was added, the reactant were heated to melt in the oil bath, and dehydrated for 1 h at 180 °C, until the volume of dehydration and the amount of the basic theory of dehydration were equal. Then the reaction system was vaccumized and controlled below 6.5 Pa, followed by heating to 220 °C and maintaining for about 1 h, and the expected product were obtained. The products were purified by the dissolution-precipitation method with CHCl3, vacuum drying for 24 h. Modified copolyesters were synthesized in the same way. P(BS-co-CHDMS) [SA:(BDO + CHDM) = 1:1.1 in molar ratio] and P(BS-co-BCHDA) [BDO:(SA + CHDA) = 1.1:1 in molar ratio] were prepared by the same method as mentioned above.
:BDO (mol:mol = 1:1.1) were added into the three-necked flask with nitrogen flow, then Ti(OiPr)4 was added, the reactant were heated to melt in the oil bath, and dehydrated for 1 h at 180 °C, until the volume of dehydration and the amount of the basic theory of dehydration were equal. Then the reaction system was vaccumized and controlled below 6.5 Pa, followed by heating to 220 °C and maintaining for about 1 h, and the expected product were obtained. The products were purified by the dissolution-precipitation method with CHCl3, vacuum drying for 24 h. Modified copolyesters were synthesized in the same way. P(BS-co-CHDMS) [SA:(BDO + CHDM) = 1:1.1 in molar ratio] and P(BS-co-BCHDA) [BDO:(SA + CHDA) = 1.1:1 in molar ratio] were prepared by the same method as mentioned above.The molecular weight and molecular weight distribution were determined by a Waters gel permeation chromatography HT3-515 (GPC, Waters Corporation U.S.) equipped with a refractive index (RI). Chloroform was used as solvent at a flow rate of 1.0 ml min−1 and 20 μl of 1.0 w/v% solution were injected for each analysis. Calibration was accomplished with polystyrene standards (Shodex, Japan).
Thermogravimetric analysis (TGA) was carried out with a TA Instruments (Q500, TA Instruments). Samples (6 ± 0.2 mg) were placed in alumina crucibles. An empty alumina crucible was used as reference. Samples were heated from ambient temperature to 500 °C in a 50 ml min−1 flow of N2. Heating rates of 10 °C min−1 were used and continuously recorded for the sample data.
MALDI-TOF-MS (Bruker Daltonics Company in USA) was used to analyze the degradation products. α-Cyano-4-hydroxycinnamic acid (α-CHCA) was used for matrix.
:1.1. For these reasons, they were used as ligands terminated by diol in the study. PBS, (PBS-co-BCHDA) and (PBS-co-CHDMS) used in the study were structurally similar to Fig. 2, respectively. Energy minimization was performed on the ligands before docking using ChemOffice v 8.0 with MM2 force field (Cambridge Company, USA).
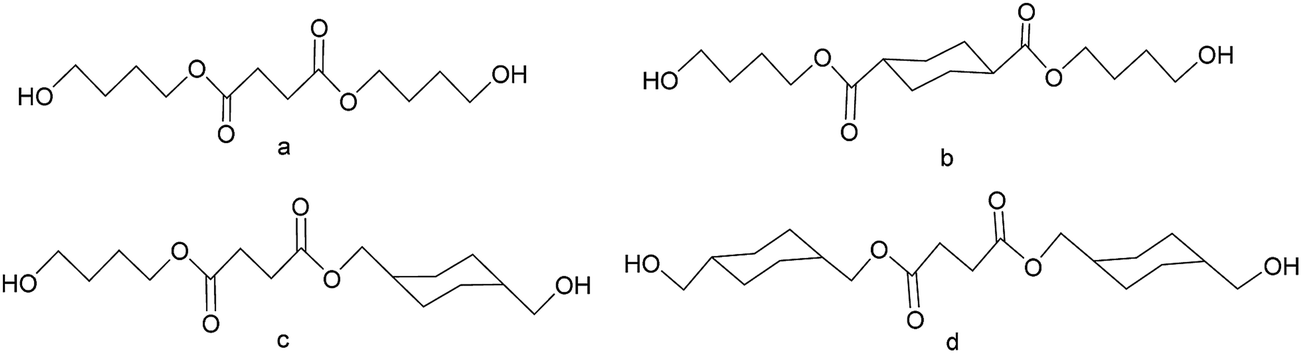 | ||
| Fig. 1 Schematic representation of: (a) BSB, (b) BCAB, (c) BSCM and (d) CMSCM, respectively. | ||
 | ||
| Fig. 2 1H-NMR spectrum of random copolymer: (A) P(BS-co-CHDMS); (B) P(BS-co-BCHDA). | ||
Results and discussion
Synthesis and characterization of the copolymers
A series of P(BS-co-CHDMS) copolyesters as well as P(BS-co-BCHDA) were synthesized by condensation polymerization of appropriate monomer feeds. For the sake of improvement of degradation of PBS, monomers with six-membered alicyclic group were introduced to designate the copolymers as shown in Table 1. PBS homopolymer was also synthesized for comparison.| Run | Polymer | BDO/CHDM or SA/CHDA in feed | BDO/CHDM or SA/CHDA in polymera | Mnb kDa | Mwb kDa | PDIb |
|---|---|---|---|---|---|---|
| a The [BDO]/[CHDM] or [SA]/[CHDA] molar ratios in polymers were determined by 1H NMR.b Obtained by GPC calibrated by polystyrene standards. | ||||||
| 1 | Pure PBS | — | — | 64.9 | 121.4 | 1.87 |
| 2 | P(BS-co-5%CHDMS) | 95/5 | 95.6/4.4 | 65.6 | 131.2 | 2.0 |
| 3 | P(BS-co-10%CHDMS) | 90/10 | 90.3/9.7 | 63.9 | 138.0 | 2.16 |
| 4 | P(BS-co-15%CHDMS) | 85/15 | 85.9/14.1 | 50.1 | 105.2 | 2.1 |
| 5 | P(BS-co-20%CHDMS) | 80/20 | 80.8/19.2 | 59.3 | 114.4 | 1.93 |
| 6 | P(BS-co-5%BCHDA) | 95/5 | 95.2/4.8 | 50.4 | 96.8 | 1.92 |
| 7 | P(BS-co-10%BCHDA) | 90/10 | 90.0/10.0 | 53.9 | 118.6 | 2.2 |
| 8 | P(BS-co-15%BCHDA) | 85/15 | 85.8/14.2 | 56.6 | 119.4 | 2.11 |
| 9 | P(BS-co-20%BCHDA) | 80/20 | 80.4/19.6 | 58.7 | 110.4 | 1.88 |
Table 1 presented the molecular characteristics of the resulting polymers. Every GPC curve showed only one peak and gave moderate molecular weight distribution. The molecular weight of the copolyesters was in the range of 50–66 kDa. The chemical compositions of the copolymers were determined from 1H NMR analysis. The typical 1HNMR spectrum of P(BS-co-CHDMS) was shown in Fig. 2A with the assignments of δ: 1.73 and 4.14 (CH2C and CH2O of BDO units linked to BS units), 2.58 (CH2CO of SA units), 3.92 (CH2O of CHDM units linked to CHDMS units), and 0.99–1.83 (CH2 and CH of CHDM units linked to CHDMS units) ppm, while P(BS-co-BCHDA) was shown in Fig. 2B with the assignments of δ: 2.58 (CH2CO of SA units linked to BS units), 1.73 and 4.14 (CH2C and CH2O of BDO units), 2.49 (CHCO of CHDA units linked to BCHDA units) and 1.46–2.18 (CH2 of CHDA units linked to BCHDA units) ppm. That was to say, the copolymers were the target products. Appropriate mechanical properties of biomaterials were essential. Our previous work showed that the tensile strength of PBS modified by the monomers with six-membered alicyclic group increased significantly, while it was widely studied. This was the reason we use the copolyesters for rapid degradation of recycling in the following studies.
GPC analysis
To the best of our knowledge, there were five kinds of lipases found to be effective in accelerating the degradation of copolyesters in organic solvent: M. miehei lipase,14 Rhizopus delemar lipase,14 N435,8,16,17 Candida rugosa lipase,14,16,17 and lipolase,8,17 all of which were obtained from fungus or bacteria. In this study, PCL was firstly found effective to degrade PBS and its copolyesters in organic solvent.The enzymatic degradation of P(BS-co-CHDMS) and P(BS-co-BCHDA) was investigated to comparison with PBS. The data of Molecular weight and molecular weight distribution were collected before and after various degradation times. As shown in Fig. 3 and 4, molecular weight of polyesters significantly decreased with the time, while the molecular weight distribution increased in the presence of PCL in CHCl3. It indicated that PBS modified by monomers with six-membered alicyclic group could obtain the optimized degradation. It was worthy of mentioning that we didn't find appreciable change of molecular weight and molecular weight distribution after 60 h without PCL. Therefore, PCL played an important role in this enzymatic degradation. Both P(BS-co-CHDMS) and P(BS-co-BCHDA) degraded much faster than PBS in CHCl3. Among the two types of copolyesters, because of introduction of monomers with six-membered alicyclic group, the regularity of helical chain structure in PBS37–40 was destroyed (Fig. 5), resulting in more space between molecular chains and more easily to rotate to fit the active pocket of PCL in chloroform. Then, the chains of polyesters were rotatable, made the ester bonds easy to exposure, which was beneficial for enzymes attacking.
 | ||
| Fig. 3 The Mn of copolyesters with different composition in the progress of degradation: (A) enzymatic degradation; (B) hydrolysis. | ||
 | ||
| Fig. 4 The molecular weight distribution of copolyesters with different composition in the progress of degradation. (A) Enzymatic degradation; (B) hydrolysis. | ||
 | ||
| Fig. 5 Schematic representation of the polymer model. | ||
After enzymatic degradations by PCL, in order to evaluate the biodegradability of the copolyesters under investigation, the degradation rates of both PBS and copolyesters with different composition were calculated (Fig. 6). We found that PBS copolymerizated with both CHDA and CHDM could improve the degradation in the CHCl3. The degradation of copolyesters first increased with the content of modified monomers, then decreased, finally kept on a certain level. This result indicated that introduction of cyclic structure in the main chain of the PBS broke its spiral structure, leading the improvement of affinity between PCL and polyester. However the larger content of modified monomer in the main chain of polyesters was, the larger steric hindrance formed by cyclic structure, which would block the combination of enzyme and substrate in the process of enzymatic degradation. Interestingly, PBS modified by CHDM had a faster degradation rate and higher degradation. Maximum degradation was observed in P(BS-co-10%CHDMS) for 85% while only 33% for P(BS-co-10%BCHDA) and 18% for pure PBS. PBS modified by CHDA, which showed high similarity to CHDM, resulted in a low degradation. The reason for this phenomenon would be investigated using molecular modeling and presented later.
 | ||
| Fig. 6 Schematic representation of the degradation. | ||
Thermogravimetric analysis (TGA)
Thermal stability of a series of synthesized copolyesters was studied by TGA before and after enzymatic degradation, as well as the pure PBS. Fig. 7 showed TGA curves of P(BS-co-CHDMS) and P(BS-co-BCHDA) samples under nitrogen atmosphere at a heating rate of 10 °C min−1. As could be seen, for P(BS-co-CHDMS), as well as the P(BS-co-BCHDA), all samples presented higher thermal stability before degradation than after degradation. These could be the reason that copolyesters had degraded to a certain extent by the catalysis of the PCL. The temperature of decomposition 5% and 15% samples were shown in Table 2. After 60 h degradation, the decomposition temperature 5% and 15% of P(BS-co-CHDMS) samples first decreased with the content of modified monomer, then increased, the same as the P(BS-co-BCHDA) samples. Furthermore, the temperature of decomposition 5% and 15% of P(BS-co-CHDMS) samples all showed lower than P(BS-co-BCHDA) with the same content saturated cyclic monomer. Obviously, PBS copolymerized with CHDM could actuate the enzymatic degradation of PBS chain, while the effect of CHDA on improvement of degradation of PBS was inconspicuous. These results further confirmed that the introduction of monomer with alicyclic group in the main chain of the PBS broke its spiral structure, leading the improvement of affinity between PCL and polyester. However, when the modified monomer content were lager, the steric hindrance formed by the cyclic structure increased, and blocked the process of enzymatic degradation. This conclusion was agreed with the result of the GPC analysis. | ||
| Fig. 7 TGA curves of copolyester samples with different composition before and after degradation: (A) P(BS-co-CHDMS); (B) P(BS-co-BCHDA). (BD: before degradation, AD: after degradation). | ||
| Poymer | Before degradation | Degradation in CHCl3 | ||
|---|---|---|---|---|
| Td-5%a/°C | Td-15%b/°C | Td-5%a/°C | Td-15% b/°C | |
| a Decomposition of 5% sample.b Decomposition of 15% sample. | ||||
| Pure PBS | 361.65 | 384.10 | 329.26 | 343.07 |
| P(BS-co-5%CHDMS) | 314.11 | 339.12 | 215.19 | 324.54 |
| P(BS-co-10%CHDMS) | 313.85 | 337.51 | 208.62 | 306.16 |
| P(BS-co-15%CHDMS) | 307.53 | 335.45 | 280.43 | 327.05 |
| P(BS-co-20%CHDMS) | 312.03 | 339.53 | 283.50 | 329.54 |
| P(BS-co-5%BCHDA) | 305.78 | 333.01 | 294.85 | 333.88 |
| P(BS-co-10%BCHDA) | 311.03 | 336.27 | 225.91 | 327.64 |
| P(BS-co-15%BCHDA) | 320.20 | 344.73 | 300.07 | 341.59 |
| P(BS-co-20%BCHDA) | 333.08 | 354.25 | 288.05 | 351.86 |
MALDI-TOF MS analysis
The enzymatic degradation of copolyester seemed to mainly proceed by internal cleavage involving a number of changes. Hydrolysis and enzymatic degradation occurred simultaneously, resulting in the formation of oligomers and reduction in molecular weight. We hypothesized that the enzymatic degradation of copolyesters should be mainly influenced by the structures. To demonstrate this hypothesis, oligomers generated during enzymatic degradation would be further characterized and analyzed.Purified products of copolyester were analyzed by MALDI-TOF MS. As the MS spectrums of the degradation products shown in Fig. 8, it gave the mass charge ratio (m/z) of ionized oligomers. After enzymatic degradation in CHCl3 with 60 h, the PBS modified by CHDA and CHDM could generate lots of oligomers, indicating that the introduction of the third component in PBS main chain could significantly improve the degradability. The degradation products were oligomers such as trimmers, tetramer, pentamer and hexamer, copolymerized by BDO, SA, CHDA and CHDM. The monomers such as BDO, SA, CHDA and CHDM could not be detected since it was not possible to detect ion fragments with molecular weights smaller than 500 g mol−1. The exact molecular forms of each oligomer from copolyesters with different composition studied were presented in Table 3.
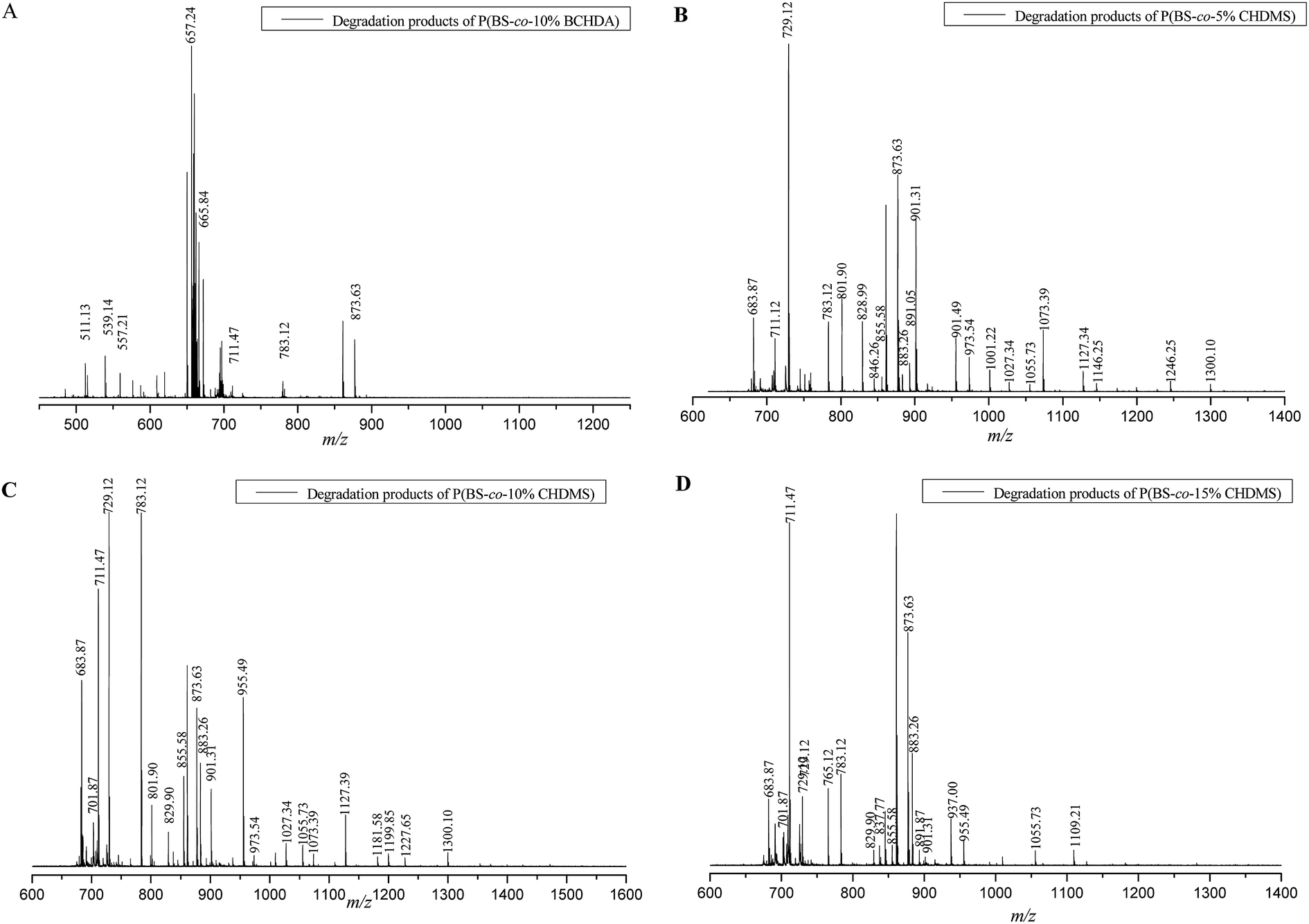 | ||
| Fig. 8 MALDI-TOF MS spectrum of the degradation products of: (A) P(BS-co-10%BCHDA); (B) P(BS-co-5%CHDMS); (C) P(BS-co-10%CHDMS); (D) P(BS-co-15%CHDMS). | ||
| Sample | a | b | c | d | |
|---|---|---|---|---|---|
| Product | [m/z]e | IRf | IRf | IRf | IRf |
| a P(BS-co-5%CHDMS).b P(BS-co-10%CHDMS).c P(BS-co-15%CHDMS).d P(BS-co-10%BCHDA).e m/z = ion mass.f Relative intensity.g Linear.h Cyclic. | |||||
| Lg (BS) (BCA)B·Na+ | 511.13 | — | — | — | 237 |
| Ch (BS)3·Na+ | 539.14 | — | — | — | 300 |
| Lg (BS)3·Na+ | 557.21 | — | — | — | 174 |
| Lg (BS)2(BCA)·Na+ | 611.89 | — | — | — | 142 |
| Lg S(BS)3·Na+ | 657.97 | — | — | — | 2486 |
| Lg (BS) (BCA)2·Na+ | 665.84 | — | — | — | 1096 |
| Lg (BS)2(CMS)B·Na+ | 683.87 | 701 | 1084 | 420 | — |
| Lg (BS)3CA·Na+; Ch (CMS)3·Na+ | 701.87 | — | 253 | 212 | — |
| Lg (BS)3CA·Na+; Lg S(BS)2(CMS)·Na+; Ch (BS)4·Na+ | 711.47 | 510 | 1625 | 2204 | 79 |
| Lg (BS)4·Na+ | 729.12 | 748 | 2072 | 450 | — |
| Lg S(BS) (CMS)2·Na+; Ch (BS)3(CMS)·Na+ | 765.12 | — | — | 497 | — |
| Lg (BS)3(CMS)·Na+; Lg (BS)3(BCA)·Na+ | 783.12 | 681 | 2067 | 592 | 111 |
| Lg (BS)4B·Na+ | 801.99 | 906 | 359 | — | — |
| Lg (BS)4S·Na+ | 829.90 | 675 | 201 | 106 | — |
| Lg (BS)2(CMS)2·Na+ | 837.77 | — | — | 135 | — |
| Lg (CMS)3CM·Na+ | 846.26 | 120 | — | — | — |
| Lg (BS)4CM·Na+ | 855.73 | 145 | 532 | 135 | — |
| Ch (BS) (BCA)3·Na+; Ch (BS) (CMS)3·Na+; Ch (BS)5·Na+ | 873.63 | 2048 | 926 | 1504 | 413 |
| 883.31 | 159 | 606 | 722 | — | |
| Lg (BS) (CMS)3·Na+ | 891.87 | 272 | — | 106 | — |
| Lg (BS)5·Na+ | 901.31 | 1660 | 458 | 58 | — |
| Lg S(BS)2(CMS)2·Na+ | 937.00 | — | — | 295 | — |
| Lg (BS)4(CMS)·Na+ | 955.49 | 510 | 989 | 159 | — |
| Lg (BS)5B·Na+ | 973.54 | 324 | 69 | — | — |
| Lg S(BS)5·Na+ | 1001.22 | 212 | — | — | — |
| Lg (BS)5CM·Na+ | 1027.34 | 86 | 138 | — | — |
| Lg S(BS)4(CMS)·Na+ | 1055.73 | 66 | 127 | 100 | — |
| Lg (BS)6·Na+ | 1073.39 | 596 | 69 | — | — |
| Ch (BS)5(CMS)·Na+ | 1109.21 | — | — | 100 | — |
| Lg (BS)5(CMS)·Na+ | 1127.39 | 198 | 306 | — | — |
| Lg (BS)4(CMS)2·Na+ | 1181.35 | — | 53 | — | — |
| Lg (BS)6CM·Na+ | 1199.09 | — | 74 | — | — |
| Lg S(BS)5(CMS)·Na+ | 1227.65 | — | 53 | — | — |
| Lg (BS)7·Na+ | 1246.25 | 106 | — | — | — |
| Lg (BS)5(CMS)B·Na+ | 1299.10 | 66 | 80 | — | — |
As can be seen in Table 3, most oligomers with BS, BCA and CMS units were generated in all samples after 60 h enzymatic degradation. The presence of oligomers were attributed to LS(BS)3·Na+, L(BS) (BCA)2·Na+, L(BS)2(CMS)B·Na+, L(BS)3(CMS)·Na+ and so on, indicated that the PCL could not only recognize the ester bond in BS style, but also in CMS and BCA style. The kinds of oligomer produced by P(BS-co-CHDMS) were more than P(BS-co-BCHDA), especially those oligomers showed high intensity in MALDI-TOF MS. Obviously, the enzymatic degradation of P(BS-co-CHDMS) was more easy than P(BS-co-BCHDA), which agreed with the GPC and TGA data. Identified in all P(BS-co-CHDMS) samples, most oligomers, such as C(BS) (CMS)3·Na+, L(BS)4(CMS)·Na+, L(BS)2(CMS)B·Na+, L(BS)3(CMS)·Na+ etc., contained CMS unit, suggesting that the introduction of the third monomer such as CHDM in PBS main chain could make the recognition of ester bond by PCL more easy, and lead to a faster degradation. It is easy to see that the enzymatic degradation of copolyesters with different structure is somewhat different from each other. All the objective data fully proved the hypothesis we proposed. It was worth noting that degradation of copolyesters produced not only linear oligomers, such as L(BS) (BCA)2·Na+, L(BS)2(CMS)B·Na+ and L(BS)3(CMS)·Na+, but also cyclic oligomers like C(BS) (CMS)3 Na+, C(BS)4·Na+and C(BS)5·Na+. Because of the catalysis of the PCL, cyclization reactions occurred in the intramolecular and intermolecular oligomer fragments that degraded from main chain of copolyesters. Similar results were also reported in our earlier experiments (Table 4).10
| Enzyme | Ligand | Ebindinga (kcal mol−1) | Einter-molb (kcal mol−1) | Evdwc (kcal mol−1) | Eelecd (kcal mol−1) | Etotale (kcal mol−1) | Etorsionalf (kcal mol−1) |
|---|---|---|---|---|---|---|---|
| a Binding energy.b Intermolecular energy.c van der Waals energies.d Electrostatic interactions.e Total energy of the complex.f Torsional free energy. | |||||||
| PCL | BSB | −5.75 | −10.23 | −9.86 | −0.36 | −1.19 | 4.47 |
| BCAB | −6.14 | −10.31 | −9.93 | −0.38 | −0.84 | 4.18 | |
| BSCM | −7.71 | −11.88 | −11.66 | −0.23 | −1.43 | 4.18 | |
| CMSCM | −9.20 | −13.08 | −12.81 | −0.27 | −0.95 | 3.88 | |
Analysis of degradation using molecular modeling
In order to explain the difference of enzymatic degradation of PBS, P(BS-co-BCHDA) and P(BS-co-CHDMS) by PCL, it was necessary to explore the binding model between enzyme and compounds, which could be considered as an alternative way to the investigation of enzymatic degradation of polyesters from molecular level. Hitherto there was, however, no research of enzymatic degradation of PBS-based copolyesters having been reported from molecular level by molecular modeling.After equilibrating PCL in organic solvents using molecular dynamics simulation, four substrate molecules were docked to the average structure PCL to determine the preferred substrate on the lipase. Binding free energy (BE), total energy (Etotal), electrostatic interactions (Eelec) and van der Waals energies (Evdw) between PCL and ligands were calculated on the basis of force field energy calculations and listed in Table 3, which showed the lowest energy ranked results. The lower binding free energy was, the steadier complex of PCL and compound was. The binding energy of PCL and compound, to some extent, represents the effectiveness of the enzymatic degradation of polyesters. From the docking simulation the observed binding free energies of the complex PCL-BCAB, PCL-BSCM and PCL-CMSCM were calculated to be −6.14, −7.71 and −9.20 kcal mol−1 respectively, which were obviously lower than the PCL-BSB free energy of binding (−5.75 kcal mol−1).
However, BSB, BCAB, BSCM and CMSCM were most likely bound to the same site located in hydrophobic pocket of subdomain of PCL. As revealed in Fig. 9, hydrogen bonding was observed between the model compounds and the residues at active site, such as Ser87. The carbonyl of the ester bond in copolyester was bonded to the Leu17 and Gln88 by strongly hydrogen bonding interaction at the active site of PCL, leading to a steady process of enzymatic degradation of polyesters. Because of the introduction of monomers with six-membered alicyclic group in PBS, like P(BS-co-BCHDA) and P(BS-co-CHDMS), the hydrophobic interactions between hydrophobic pocket of PCL and compounds became stronger, leading to a more stable docking in active site and better degradation. It was worth to pay attention that due to rigid structure of six-membered alicyclic group in acid segment, BCAB couldn't rotate to fix the active pocket of PCL, leading to a collision between BCAB and the side wall of the active pocket (Fig. 10). Because of the collision between BCAB and the side wall of the active pocket in PCL, BCAB couldn't reach the bottom of the active pocket and interact with the residues strongly at the active site of PCL. When it docked to PCL, it would result in a longer distance between the carbonyl of BCAB and Leu17 and Gln88 and less stability than CMSCM. In other words, the progress of enzymatic degradation of P(BS-co-BCHDA) catalyzed by catalytic triad of Ser87, Asp264 and His286, could be blocked by the structure with alicyclic group in acid segment. Compared with P(BS-co-BCHDA), the acid segments in the polymer chains of P(BS-co-CHDMS) were all SA, which could allow P(BS-co-CHDMS) to rotate to fit the active pocket and dock steadily (Fig. 10). These results indicated that the enzymatic degradation of the polyesters by PCL became increasingly susceptible to the modification by monomers with six-membered alicyclic group, which could destroy the compact rigid structure of PBS, make the combination of enzyme and substrate easier and finally accelerate of degradation. However, the effect of copolymerization by CHDM seemed to be better than CHDA on improvement of enzymatic degradation of PBS. Due to the steric hindrance formed by the cyclic structure in acid segment with the CHDA, the enzymatic degradation of P(BS-co-BCHDA) was severely inhibited.
 | ||
| Fig. 9 Three dimensional view of the interactions between the active site residues of PCL and the top-ranking docked conformations of compounds: (A) BCAB (light blue), (B) (carbon atoms in pink). The hydrogen bond between the active site residues and ligand is depicted in yellow dashed line. | ||
 | ||
| Fig. 10 The surface of PCL with the top-ranking docked conformation of compounds BCAB (light brown) and CMSCM (carbon atoms in light blue). | ||
Conclusions
This study suggested that a novel degradation process could be developed for biodegradable materials using PCL in CHCl3, paving a road for new solution to rapid recycling carbon resources. A series of copolyesters based on the biodegradable PBS matrix and pure PBS were prepared by melt polycondensation and used to investigate the biodegradation in chloroform at 55 °C for 60 h. In terms of results, all the copolyesters degraded faster than pure PBS, while the degradation rates were higher in the presence of PCL in CHCl3. These results suggested the introduction of monomers with six-membered alicyclic group could break the rigid spiral structure of PBS, enhanced the stability of the combination between copolyesters and PCL. Major products of the enzymatic degradation were linear and cyclic oligomers, which contained the units of BCHDA or CHDMS, indicating that the monomers with six-membered alicyclic group in the main chain of copolyesters could make the recognition of ester bond by PCL easier and accelerate the enzymatic degradation.The degradation of polyesters in CHCl3 was in the order of P(BS-co-CHDMS) > P(BS-co-BCHDA) > PBS. It seemed that the PBS modified by CHDM showed better effect than that by CHDA on the improvement of enzymatic degradation. The molecular modeling shed light on the interactions between the residues at the active site of PCL and polyester. The results indicated that the difference of enzymatic degradation of P(BS-co-CHDMS) and P(BS-co-BCHDA) was mainly attributed to the structures of polyesters. Due to the steric hindrance formed by CHDA in acid segment of P(BS-co-BCHDA), the substrates were blocked to reach the bottom of the active pocket, made the combination between the carbonyl of ester group and the Leu17 and Gln88 unstable. Then, the progress of enzymatic degradation was inhibited.
Acknowledgements
We acknowledge the financial support from the National High Technology Research and Development Program of China (2011AA100503) and Specialized Research Fund for the Doctoral Program of Higher Education (20126125110001).Notes and references
- M. Nerantzaki, G. Z. Papageorgiou and D. N. Bikiaris, Polym. Degrad. Stab., 2014, 108, 257–268 CrossRef.
- J. C. Middleton and A. J. Tipton, Biomaterials, 2000, 21, 2335–2346 CrossRef CAS PubMed.
- A. L. Carbone and K. E. Uhrich, Macromol. Rapid Commun., 2009, 30, 1021–1026 CrossRef CAS PubMed.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef CAS PubMed.
- N. Kukreja, Y. Onuma, J. Daemen and P. W. Serruys, Pharmacol. Res., 2008, 57, 171–180 CrossRef CAS PubMed.
- J. Venugopal and S. Ramakrishna, Tissue Eng., 2005, 11, 847–854 CrossRef CAS PubMed.
- H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Prog. Polym. Sci., 2012, 37, 237–280 CrossRef CAS.
- G. Sivalingam, S. Chattopadhyay and G. Madras, Polym. Degrad. Stab., 2003, 79, 413–418 CrossRef CAS.
- S. Kobayashi, H. Uyama and T. Takamoto, Biomacromolecules, 2000, 1, 3–5 CrossRef CAS PubMed.
- M. Zhang, M. L. Ding, T. Zhang and J. M. Yang, Chem. J. Chin. Univ., 2010, 31, 612–615 CAS.
- S. Okajima, R. Kondo, K. Toshima and S. Matsumura, Biomacromolecules, 2003, 4, 1514–1519 CrossRef CAS PubMed.
- Z. Gan, D. Yu, Z. Zhong, Q. Liang and X. Jing, Polymer, 1999, 40, 2859–2862 CrossRef CAS.
- H. Shirahama, Y. Kawaguchi, M. S. Aludin and H. Yasuda, J. Appl. Polym. Sci., 2001, 80, 340–347 CrossRef CAS.
- L. Pastorino, F. Pioli, M. Zilli, A. Converti and C. Nicolini, Enzyme Microb. Technol., 2004, 35, 321–326 CrossRef CAS.
- S. Matsumura, H. Ebata and K. Toshima, Macromol. Rapid Commun., 2000, 21, 860–863 CrossRef CAS.
- G. Sivalingam, S. Chattopadhyay and G. Madras, Chem. Eng. Sci., 2003, 58, 2911–2919 CrossRef CAS.
- G. Sivalingam and G. Madras, J. Appl. Polym. Sci., 2004, 91, 2391–2396 CrossRef CAS.
- T. Takamoto, H. Shirasaka, H. Uyama and S. Kobayashi, Chem. Lett., 2001, 492–493 CrossRef CAS.
- D. C. Rapaport. The art of molecular dynamics simulation. Cambridge University Press, 2004 Search PubMed.
- X. Sun, H. Ågren and Y. Tu, J. Phys. Chem. B, 2014, 118, 14737–14744 CAS.
- X. Sun, H. Ågren and Y. Tu, J. Phys. Chem. B, 2014, 118, 10863–10873 CrossRef CAS PubMed.
- X. Sun, J. Cheng and X. Wang, et al., Sci. Rep., 2015, 5, 8066 CrossRef CAS PubMed.
- H. Zhou, Y. Qu and Y. Bu, et al., ChemPlusChem, 2012, 77, 293–300 CrossRef CAS.
- H. Zhou, Y. Qu and C. Kong, et al., Appl. Microbiol. Biotechnol., 2013, 97, 10399–10411 CrossRef CAS PubMed.
- M. Zhang, S. Lai and J. Song, Chem. J. Chin. Univ., 2008, 29, 1246–1249 Search PubMed.
- M. Luic, Z. Štefanic and I. Ceilinger, et al., J. Phys. Chem. B, 2008, 112, 4876–4883 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- A. K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P. C. Nair, C. Oostenbrink and A. E. Mark, J. Chem. Theory Comput., 2011, 7, 4026–4037 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. J. Pedersen, Chem. Phys., 1995, 103, 8577–8593 CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, Autodock, version 4.2.5, The Scripps Research Institute, La Jolla, CA, USA, 2013 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- W. L. DeLano, The PyMOL Molecular Graphics System, Revision 0.99, DeLano Scientific, San Carlos, CA, USA, 2002 Search PubMed.
- Y. Ichikawa, H. Kondo and Y. Igarashi, et al., Polymer, 2000, 41, 4719–4727 CrossRef CAS.
- Y. Ichikawa, J. Suzuki and J. Washiyama, et al., Polymer, 1994, 35, 3338–3339 CrossRef CAS.
- C. Y. Li, D. Yan and S. Z. D. Cheng, et al., Macromolecules, 1999, 32, 524–527 CrossRef CAS.
- E. Yashima, K. Maeda and H. Iida, et al., Chem. Rev., 2009, 109, 6102–6211 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |