DOI:
10.1039/C5RA13275D
(Paper)
RSC Adv., 2016,
6, 2632-2640
Photo-reduction assisted synthesis of MnO2/reduced graphene oxide/P25 for electrochemical detection of hydrogen peroxide
Received
7th July 2015
, Accepted 17th December 2015
First published on 22nd December 2015
Abstract
In this work, we report the synthesis of MnO2/RGO (reduced graphene oxide)/P25 nanocomposites for a non-enzymatic hydrogen peroxide sensor. MnO2/RGO/P25 nanocomposites were synthesized with a photo-reduction approach and characterized by field emission scanning electron microscopy, X-ray photoelectron spectroscopy, Fourier transform infrared spectroscopy and Raman spectroscopy. A non-enzymatic hydrogen peroxide sensor was fabricated by dropping MnO2/RGO/P25 nanocomposites on the surface of a glassy carbon electrode. Electrochemical measurements of the MnO2/RGO/P25 modified electrode were carried out on an electrochemical workstation. The as-prepared sensor exhibited high electrocatalytic activity, selectivity and stability towards the oxidation of H2O2. Under optimum conditions, the calibration curve for H2O2 determination was linear in the range from 1.0 × 10−6 to 4.0 × 10−3 M (R2 = 0.999) with a detection limit of 3 × 10−7 M (S/N = 3).
1. Introduction
Hydrogen peroxide is one of the most important and common oxidizing agents widely utilized in clinical areas, biochemistry, the pharmaceutical industry and environmental fields.1 Therefore, accurate and fast detection of H2O2 via highly sensitive and cost-effective sensors is of great practical significance. Thus, many detection methods have been developed, such as titrimetry,2 chemiluminescence,3–4 fluorescence,5 spectrophotometry6 and the electrochemical method.7–9 Among them, the electrochemical method is the most attractive because of its high sensitivity, low cost and operational simplicity. Over the past decades, intensive interests have been directed to electrochemical detection of H2O2 based on enzymes due to their high sensitivity and selectivity.10–12 However, the drawbacks of enzymes based sensors such as limited lifetime, instability, poor reproducibility and complicated fabrication procedures have impeded them from being prevailing.13 In recent years, much effort has been devoted to non-enzymatic electrochemical detection of H2O2 to overcome these disadvantages due to the intrinsic fragility nature of enzymes.14 Various materials have been successfully applied extensively in fabricating high efficient non-enzymatic electrochemical sensors, such as metals,15–18 alloys,19 transition metal oxide nanoparticles20 and graphene.21–23 In particular, as one outstanding electrocatalytic material towards H2O2, manganese dioxide has drawn intensive attentions in virtue of excellent physicochemical properties, low cost, environmental benignity, safe operation voltage and non-toxic.24–26
Reduced graphene oxide (RGO) is a kind of attractive material in the scientific community owning to its excellent mobility of charge carriers, high thermal conductivity, great chemical stability, high mechanical flexibility and large specific surface area.27–29 So far, various synthesis methods have been developed to prepare RGO such as chemical reduction, electrochemical reduction and thermal reduction methods.30–33 In recent years, photo-reduction methods turning graphene oxide into RGO in the presence of TiO2 and avoiding drawbacks caused by chemical reduction methods and thermal reduction methods have been developed by researchers.34 Compared with the chemical and thermal reduction of GO, photo-reduction is a facile and green reduction method to prepare RGO.
TiO2 is one of the most promising materials and has been well studied over the years because of its photocatalytic properties, non-toxicity, high chemical stability and huge potential application value.35–37 When TiO2 is irradiated with UV lights, the electrons will be excited from the valence bands to the conduction bands, leaving holes in valence bands and generating photo electron–hole pairs in this process.38 Photogenerated electrons and holes migrate on surface of TiO2, triggering reductive reaction and oxidative reaction, respectively. P25 is a kind of commercial TiO2 nanoparticle consisting 80% anatase phase and 20% rutile phase. The mixed phases suppress the recombination of the photo-electrons and photo-holes, which promote its photocatalytic efficiency. Based on this, many kinds of TiO2–RGO nanocomposites have been developed to improve the photocatalysis property of TiO2.
In this work, we successfully synthesized MnO2/RGO/P25 nanocomposites using photo-reduction method. MnO2/RGO/P25 nanocomposites were characterized by field emission scanning electron microscopy (FESEM), X-ray photoelectron spectroscopy (XPS), Fourier transform infrared (FTIR) spectroscopy and Raman spectroscopy. Cyclic voltammetry (CV) of MnO2/RGO/P25 modified sensor were conducted on electrochemical workstation. The as-prepared sensor exhibited high electrocatalytic activity, good selectivity and stability toward the oxidation of H2O2. To the best of our knowledge, this is the first report on photo-reduction assisted synthesis of MnO2/RGO/P25 nanocomposites and their use in electrochemical detection of hydrogen peroxide.
2. Experimental section
2.1 Materials and apparatus
Graphene oxide was purchased from Institute of Coal Chemistry, Chinese Academy of Sciences (Shanxi, China). The commercial TiO2 (Degussa P25) was obtained from Degussa Co., Ltd. (Germany). KMnO4, KH2PO4, K2HPO4·3H2O, hydrogen peroxide (H2O2, 30%), CH3CH2OH and NaOH were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). 0.1 M phosphate buffer solutions (PBS, pH 6–10) were prepared from NaH2PO4, Na2HPO4·3H2O and NaOH and used as supporting electrolytes. All chemicals were of analytical grade. Ultra-pure water was used throughout the experiment. UV lamp (16 W) was purchased from Hangzhou Yaguang Lamp Co., Ltd. (Hangzhou, China).
The morphologies of GO, RGO/P25 and MnO2/RGO/P25 were characterized by field emission scanning electron microscopy (Zeiss, Germany). The chemical states of elements in MnO2/RGO/P25 were analyzed by WSCALAB 250 Xi X-ray photoelectron spectroscopy (England) using a source of Al Kα radiation with energy of 1486.6 eV. Infrared spectra of GO, RGO/P25 and MnO2/RGO/P25 were carried out on an AVATAR 370 Fourier transform infrared spectrometer (FTIR, America) in the region of 400–4000 cm−1. Raman spectra of GO, RGO/P25 and MnO2/RGO/P25 were characterized by inVia Reflex (Renishaw, England) with an excitation laser wavelength of 514.5 nm. All electrochemical measurements were performed on a CHI660C Electrochemical Workstation (Shanghai Chenhua, China) with a conventional three-electrode system, in which a saturated calomel electrode (SCE), platinum foil and MnO2/RGO/P25 modified GCE were served as the as reference, counter and working electrode, respectively.
2.2 Synthesis of MnO2/RGO/P25 nanocomposites
MnO2/RGO/P25 nanocomposites were synthesized using photo-assisted reduction method. In brief, 0.2 mg P25 was dispersed in 2 mL GO (1.0 mg mL−1) solution under magnetic stirring at room temperature (25 °C) and the suspension solution was vigorously stirred for 1 h under UV irradiation to obtain RGO/P25 homogenous dispersion. After that, a series of KMnO4 (0.2, 0.4, 0.6, 0.8, and 1.0 mg) solutions with different amount were dropwise added into the same volume RGO/P25 homogenous dispersion solutions which had been irradiated with UV light to synthesize MnO2/RGO/P25 nanocomposites. Finally, MnO2/RGO/P25 nanocomposites were centrifuged and washed with ultrawater for 3 times to further analysis.
2.3 Preparation of MnO2/RGO/P25 modified electrodes
A glassy carbon electrode (GCE, 3 mm in diameter) was polished with 0.05 μm alumina slurries using Buehler polishing kit, followed by washing with deionized water, ethanol and finally deionized water under ultrasound conditions (each for 3 min), and drying at room temperature to remove any adsorbed impurities on the electrode surface. 5 μL of as-prepared MnO2/RGO/P25 nanocomposites was dropped onto the surface of GCE and dried in air under the infrared lamp for 15 min. For comparison, 5 μL of GO and RGO/P25 were dropped onto GCE and dried at the same conditions as MnO2/RGO/P25–GCE to obtain GO–GCE and RGO/P25–GCE.
3. Results and discussion
3.1 Photo-reduction synthesis of MnO2/RGO/P25 nanocomposites
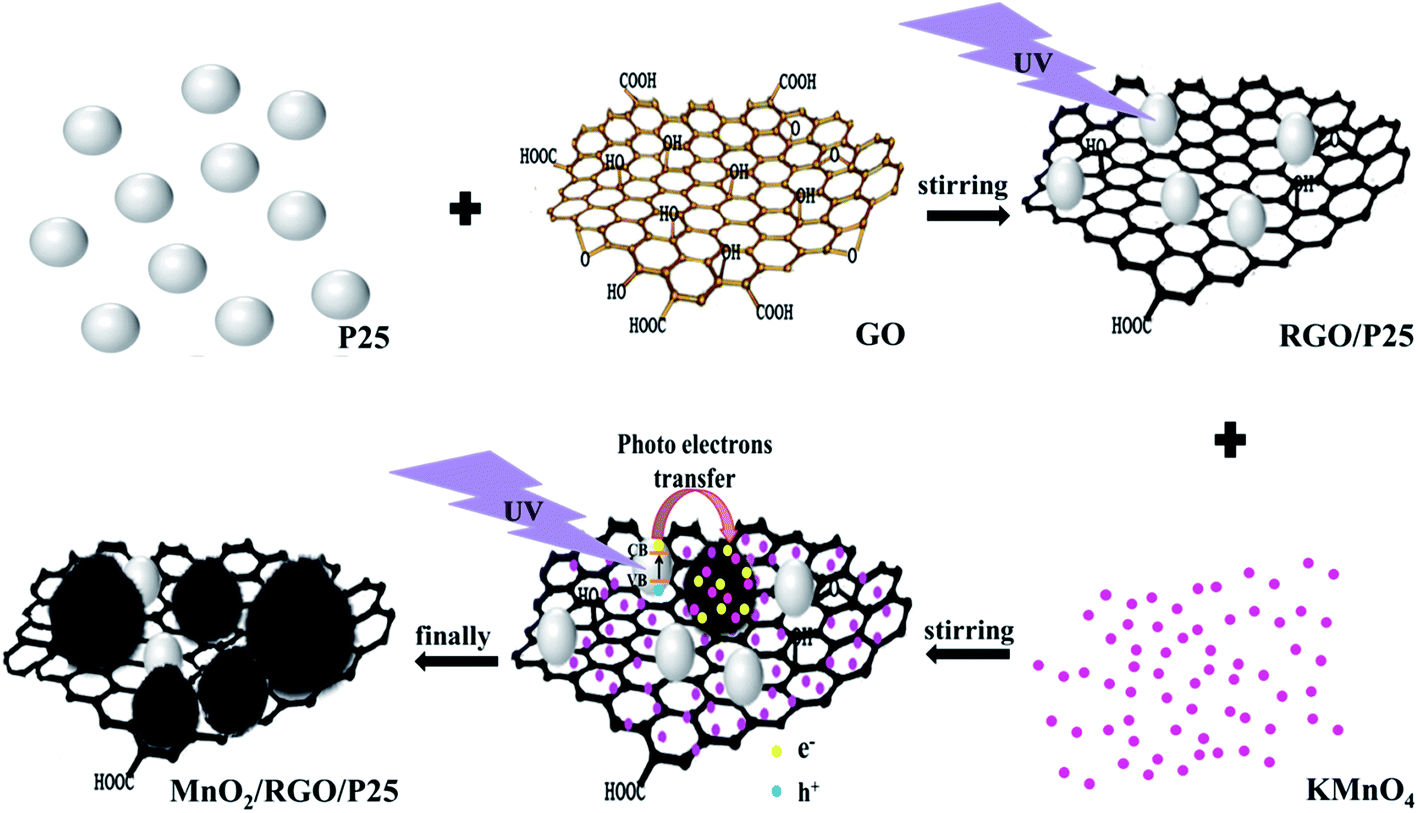
As shown in Scheme 1, when P25 was first irradiated with UV lights, the electrons would be excited from the valence bands to the conduction bands, leaving holes in valence bands and generating photo electron–hole pairs in this process (eqn (1)). Then, the photo holes reacted with H2O to generate oxygen and GO was photo-reduced to RGO with photo electrons (eqn (2) and (3)). Lastly, KMnO4 solution was drop-wise added into RGO/P25 homogenous dispersion solution irradiated with UV light to synthesize MnO2/RGO/P25 nanocomposites (eqn (4)). The photo-assisted reduction mechanism might be proposed as follows:34,39–43| | |
P25 + hν → P25 (3h+) + P25 (3e−)
| (1) |
| |
P25 (3h+) + ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) H2O → P25 + ¾O2 + 3H+ H2O → P25 + ¾O2 + 3H+
| (2) |
| | |
P25 (3e−) + GO → RGO/P25
| (3) |
| | |
RGO (3e−)/P25 + MnO4− + 4H+ → MnO2/RGO/P25 + 2H2O
| (4) |
 |
| | Scheme 1 Photo-reduction synthesis of MnO2/RGO/P25 nanocomposites. | |
3.2 Field emission scanning electron microscopy (FESEM)
Morphologies of GO, RGO/P25 and MnO2/RGO/P25 were characterized by FESEM. As shown in Fig. 1A, GO displays a tentative structure with affluent wrinkles. From the image of RGO/P25 nanocomposites (Fig. 1B), P25 nanoparticles with diameter in the range of 20–30 nm were wrapped by RGO and attached on the surface of photo-reduced graphene oxide sheets. Fig. 1C shows MnO2/RGO/P25 nanocomposites were formed after MnO4− ions were photo-reduced and anchored on the surface of RGO/P25. RGO/P25 is likely to provide the nucleation sites for growing preferably to form MnO2 nanoplatelets. Most of MnO2 nanoparticles have a size of ∼100 nm and disperse on the surface of RGO/P25 nanocomposites. Fig. 1D–F correspond to real samples of GO, RGO/P25 and MnO2/RGO/P25 composites, respectively. Obviously, the colors of GO, RGO/P25 and MnO2/RGO/P25 visibly change from brownish to black after irradiation.
 |
| | Fig. 1 FESEM images of GO (A), RGO/P25 (B) and MnO2/RGO/P25 (C); photos of GO (D), RGO/P25 (E) and MnO2/RGO/P25 (F). | |
3.3 X-ray photoelectron spectroscopy
The surface chemical states of GO, RGO/GO, MnO2/RGO/P25 were examined by XPS. Fig. 2A shows the XPS of MnO2/RGO/P25. Different C1s binding energies imply the existence of different chemical environments of carbon in GO (Fig. 2B), RGO/GO (Fig. 2C), MnO2/RGO/P25 (Fig. 2D). As shown in spectra, four peaks with binding energies of 284.8, 286.7, 287.4 and 288.9 eV, can be assigned to the contributions of the C–C (sp2) bonds, C–O bonds, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds and OC–O bonds,44 respectively. Compared to GO, peak intensity at 284.8 eV in RGO/GO and MnO2/RGO/P25 increased after illumination, indicating GO were reduced into RGO. Fig. 2E shows the Mn 2p XPS of MnO2/RGO/P25. The peaks observed at 641.9 and 653.7 eV with a BE difference of 11.8 eV were ascribed to Mn 2p3/2 and Mn 2p1/2, respectively.45 These results agree well with Mn(IV) characters in phase of MnO2.
O bonds and OC–O bonds,44 respectively. Compared to GO, peak intensity at 284.8 eV in RGO/GO and MnO2/RGO/P25 increased after illumination, indicating GO were reduced into RGO. Fig. 2E shows the Mn 2p XPS of MnO2/RGO/P25. The peaks observed at 641.9 and 653.7 eV with a BE difference of 11.8 eV were ascribed to Mn 2p3/2 and Mn 2p1/2, respectively.45 These results agree well with Mn(IV) characters in phase of MnO2.
 |
| | Fig. 2 XPS of MnO2/RGO/P25 nanocomposites (A); C1s XPS spectra of GO (B), RGO/P25 (C) and MnO2/RGO/P25 nanocomposites (D); Mn 2p XPS spectra of MnO2/RGO/P25 nanocomposites (E). | |
3.4 Raman and FTIR spectroscopy
Samples of GO, RGO/P25 and MnO2/RGO/P25 were also characterized by Raman spectroscopy recorded between 100 and 2000 cm−1 at the same laser power and acquisition time. As depicted in Fig. 3A, GO shows two broadened characteristic frequencies at 1344 and 1597 cm−1 corresponding to the D and G bands, respectively.46–47 Higher intensity ratio means higher defects in GO comprising less sp2 hybridization. The intensity ratio of D and G bands (ID/IG) of GO is 1.06. After photo-reduction reaction process, this value decreases to 1.03 in RGO/P25 and 1.02 in MnO2/RGO/P25 indicating more graphitization of GO. Compared MnO2/RGO/P25 with RGO/P25, there is no significant change of the ID/IG value after further UV-light irradiation, which indicates no obvious sp2 hybridization after further photo-reduction reaction.48 Raman results are also in agreement with the X-ray photoelectron spectra in Fig. 2. Raman spectra of RGO/P25 and MnO2/RGO/P25 further show frequencies at 398 and 507 cm−1 ascribing to the B1g and A1g vibrational modes of TiO2 anatase phase, respectively. The presence of rutile phase is characterized by peaks at 153 and 630 cm−1 which correspond to the modes B1g and A1g. The band at 680 cm−1 of MnO2/RGO/P25 is always seen in MnO2 materials and corresponds to the symmetric stretching vibration (Mn–O) of the octahedral [MnO6] groups49 indicating that MnO2 were introduced. The red shift in the G band from 1597 to 1591 cm−1 is possibly due to increased numbers of isolated carbon double bonds in disordered GO after photo-reduction reaction.
 |
| | Fig. 3 Raman spectra of GO, RGO/P25 and MnO2/RGO/P25 (A); FTIR spectra of GO, RGO/P25 and MnO2/RGO/P25 (B). | |
FTIR spectra of GO, RGO/P25 and MnO2/RGO/P25 nanocomposites were recorded in 400–4000 cm−1. As shown in Fig. 3B, all samples present a broad absorption peaks around 3300 cm−1, which are attributed to the surface absorbed water.50 In the FTIR spectrum of GO, the absorption peaks at 1732 and 1622 cm−1 correspond to the CO stretching and CC stretching vibrations, respectively (curve a). The band at 1223 cm−1 can be ascribed to the epoxy groups vibrations of GO. Compared to GO, the peaks at 1732 and 1223 cm−1 of RGO/P25 and MnO2/RGO/P25 nanocomposites disappeared, which indicate reduction of GO and its transformation into RGO (curve b and c). The peak appearing at 1053, 1070 and 1093 cm−1 are assigned to the vibrations of C–O in alkoxy of GO (curve a), RGO/P25 (curve b) and MnO2/RGO/P25 (curve c), respectively. For RGO/P25 nanocomposites, the broad absorption at low frequency of 663 cm−1 and 519 cm−1 (below 1000 cm−1) are attributed to the vibration of Ti–O–Ti bonds in P25. FTIR spectrums of MnO2/RGO/P25 nanocomposites also show two peaks at 617 and 509 cm−1 corresponding to the stretching vibrations of Mn–O–Mn and Mn–O bonds, respectively.
3.5 Electrochemical behavior of MnO2/RGO/P25 modified glassy carbon electrode
In order to evaluate the electrocatalytic properties of MnO2/RGO/P25 to oxidation of H2O2, the performance of bare GCE, GO–GCE, RGO/P25–GCE and MnO2/RGO/P25–GCE was evaluated by cyclic voltammetry in 0.1 M PBS (pH 8) at the scan rate of 100 mV s−1. Fig. 4 shows CVs of bare GCE (A), GO–GCE (B), RGO/P25–GCE (C) and MnO2/RGO/P25–GCE (D) in the absence (curve a, b, c and d) and presence (curve a′, b′, c′ and d′) of 1 mM H2O2 in 0.1 M PBS (pH 8) at scan rate of 100 mV s−1. In the absence of H2O2, no oxidation peaks were observed at bare GCE (curve a) and GO–GCE (curve b). A very weak oxidation peak around +0.61 V in the absence of 1 mM H2O2 in 0.1 M PBS (pH 8) was observed at RGO/P25–GCE (Fig. 4C, insert) which might be ascribed to oxidation of RGO.51 In contrast MnO2/RGO/P25–GCE (curve d) displayed a pair of broad but weak peaks in the potential range of 0–1.0 V in the absence of H2O2, which might be attributed to the reduction of MnO2 to Mn(II or III) and the reoxidation of Mn(II or III) back to MnO2.52–53 When 1 mM H2O2 was added into 0.1 M PBS (pH 8), no obvious electrochemical oxidation peaks were observed at bare GCE (curve a′) and GO–GCE (curve b′). A weak electrochemical oxidation peak was observed at RGO/P25–GCE (curve c′). Compared with bare GCE (curve a′), GO–GCE (curve b′) and RGO/P25–GCE (curve c′), a broad and strong oxidation peak was observed on MnO2/RGO/P25–GCE (curve d′) at around +0.7 V in 0.1 mM H2O2 + 0.1 M PBS (pH 8). Considering the standard potential values of Mn species, we can make a conclusion that reductive peaks and oxidative peaks are assigned to the reduction of MnO2 to Mn(II, III) (eqn (5) and (6)) and the oxidation of Mn(II, III) to Mn(IV) (eqn (7) and (8)),52–53 respectively. The electrochemical reaction mechanism is proposed as following equations:| | |
2MnO2/RGO/P25 (s) + H2O2 (aq) → Mn2O3/RGO/P25 (s) + O2 (g) + H2O
| (5) |
| | |
MnO2/RGO/P25 (s) + H2O2 (aq) → Mn(OH)2/RGO/P25 (s) + O2 (g)
| (6) |
| | |
Mn2O3/RGO/P25 (s) + 2OH− (aq) → 2MnO2/RGO/P25 (s) + H2O (l) + 2e−
| (7) |
| | |
Mn(OH)2/RGO/P25 (s) + 2OH− (aq) → MnO2/RGO/P25 (s) + 2H2O (l) + 2e−
| (8) |
 |
| | Fig. 4 Cyclic voltammograms of bare GCE (A), GO–GCE (B), RGO/P25–GCE (C) and MnO2/RGO/P25–GCE (D) in the absence (curve a, b, c and d) and presence (curve a′, b′, c′ and d′) of 1 mM H2O2 in 0.1 M PBS (pH 8) at scan rate of 100 mV s−1; amperometric response of bare GCE, GO–GCE, RGO/P25–GCE and MnO2/RGO/P25–GCE on a successive of 0.5 mM H2O2 added into 0.1 M PBS (pH 8) with applied potential of +0.6 V (E). Insets: the amplified cyclic voltammogram of GCE (a), GO–GCE (b), RGO/P25–GCE (c) and MnO2/RGO/P25–GCE (d) in the absence (curve c) of 1 mM H2O2 in 0.1 M PBS (pH 8). | |
The amperometric response of bare GCE, GO–GCE, RGO/P25–GCE and MnO2/RGO/P25–GCE toward a successive of 0.5 mM H2O2 were also investigated in 0.1 M PBS (pH 8) with applied potential of +0.6 V, as shown in Fig. 4E. MnO2/RGO/P25 nanocomposites modified GCE exhibited highest sensitivity.
3.6 Optimization of experimental parameters
The content of MnO2 is a key factor to influence the response current. The influence of KMnO4 was investigated using amperometric response to 0.5 mM H2O2 in 0.1 M PBS (pH 8) at the scan rate of 100 mV s−1. As shown in Fig. 5A, the response current of H2O2 increased with increase of KMnO4 from 0 to 0.6 mg. However, the response current decreased with further increase of KMnO4. At first, a large number of MnO2 can be easily anchored on the surfaces of RGO, which provide more electrocatalytic sites and significantly increase the response current towards H2O2. However, further increases of KMnO4 result in the agglomeration of MnO2, which decrease conductivity of MnO2/RGO/P25 and slows mass transport through the electrode surface.54
 |
| | Fig. 5 Effects of KMnO4 (A) and applied potential (C) on the amperometric response of 0.5 mM H2O2 in 0.1 M PBS (pH 8) at scan rate of 100 mV s−1 with applied potential of +0.6 V; effects of different pH value on the amperometric response of 0.5 mM H2O2 in 0.1 M PBS (pH 8) at scan rate of 100 mV s−1 (B). Error bars represent the standard deviation for three independent measurements. | |
The pH value of the electrolyte is also a crucial parameter affecting the stability and activity of the modified electrode. In order to obtain optimal amperometric response to H2O2, different pH values on the amperometric response of 0.5 mM H2O2 in 0.1 M PBS were also tested with the applied potential of +0.6 V at the scan rate of 100 mV s−1 Fig. 5(B). The response current of H2O2 rose strikingly upon the increase of pH 6–8 and reached the maximum for pH 8, and then sharply decreased. Thus, pH 8 was chosen as the optimal pH for amperometric H2O2 sensing.
It is well known that the applied potential strongly affects the amperometric responses. The influence of the applied potential of the electrocatalytic activity of MnO2/RGO/P25–GCE was investigated using amperometric response to 0.5 mM H2O2 in 0.1 M PBS (pH 8) at the scan rate of 100 mV s−1. The response current increased sharply with the increase of applied potential until the potential reached +0.6 V. The response current drastically decreased when the applied potential exceeded +0.6 V. Therefore, +0.6 V was selected for the amperometric detection of H2O2.
3.7 Linearity, selectivity and stability
Under the optimum conditions, MnO2/RGO/P25–GCE exhibited high rapid and sensitive response for H2O2 as displayed in Fig. 6A. The current response is linear with H2O2 concentration in the range of 1–4000 μM with sensitivity of 297.2 μA mM−1 cm−2, as shown in Fig. 6B. The corresponding regression equation is I (μA) = 0.754 + 0.021C (μM) (R2 = 0.999). The detection limit is estimated to be 0.3 μM (S/N = 3). Selectivity is also an important aspect for sensors. The interferences of various common interfering substances including 100-fold concentration inorganic species such as CO32−, NO3−, CH3COO−, Cl−, K+ and Na+ and organic species ascorbic acid (AA, 1-fold), uric acid (UA, 1-fold) and glucose (10-fold) were evaluated towards H2O2 analysis. The interferences with the amperometric determination of 0.25 mM H2O2 was implemented under optimal conditions in 0.1 M PBS (pH 8), as shown in Fig. 6C. The results show that all interfering agents had response currents are less than 4% of that produced by H2O2, indicating high selectivity of MnO2/RGO/P25–GCE for detecting H2O2. We also used the amperometric response method to evaluate the stability of MnO2/RGO/P25–GCE at +0.6 V in 0.1 M PBS (pH 8). As shown in Fig. 6D, the respond current change a little during 1000 s test period indicating that MnO2/RGO/P25–GCE shows high stability (Table 1).
 |
| | Fig. 6 Amperometric response of MnO2/RGO/P25–GCE of different concentrations of H2O2 in 0.1 M PBS (pH 8) at applied potential of +0.6 V (A); corresponding calibration curve of MnO2/RGO/P25–GCE (B); amperometric response of MnO2/RGO/P25–GCE to 0.25 mM H2O2 and different interferences in stirring 0.1 M PBS (pH 8) with applied potential at +0.6 V (C); the current change of MnO2/RGO/P25–GCE to 0.25 mM H2O2 during 1000 s test period in 0.1 M PBS (pH 8) with applied potential at +0.6 V (D). | |
Table 1 Performance comparison of the proposed MnO2/RGO/P25–GCE with other glucose sensors
| Modified electrode |
Linear range (μM) |
Detection limit (μM) |
Sensitivity (μA mM−1 cm−2) |
Reference |
| NTA stands for nitrilotriacetic acid. VACNTs stands for vertically aligned multiwalled carbon nanotubes. CF stands for carbon foam. CNT stands for graphene/carbon nanotubes. |
| Mn-NTAa/Nafion |
5–2500 |
0.2 |
78.9 |
9 |
| MnO2 nanorods |
— |
5 ± 2.5 |
— |
53 |
| GO/MnO2 |
5–600 |
0.8 |
38.2 |
55 |
| MnO2/VACNTsb |
1.2–1800 |
0.8 |
1080 |
56 |
| MnO2/CFc |
2.5–2055 |
0.12 |
54 |
57 |
| MnO2/graphene/CNTd |
1–1030 |
0.1 |
539.4 |
58 |
| MnO2/RGO/P25 |
1–4000 |
0.3 |
297.2 |
This work |
3.8 Real sample analysis
The application for real sample analysis of MnO2/RGO/P25–GCE was evaluated by the determination of H2O2 in toothpaste. Briefly, 1.0 g toothpaste mixed with 10 mL PBS (0.1 M pH 8) was centrifuged before test. H2O2 has been estimated directly by amperometric method under optimal conditions with standard addition method. The detection result is shown in Table 2. The results have demonstrated that MnO2/RGO/P25 has potential applications in the determination of H2O2 in real samples.
Table 2 Determination of H2O2 in toothpaste samples (n = 3)
| Toothpaste samples |
Detected (μM) |
Added (μM) |
Founded (μM) |
Recovery (%) |
RSD (%) |
| 1 |
18.24 |
20.00 |
38.38 |
100.3 |
2.9 |
| 2 |
20.05 |
20.00 |
38.81 |
96.7 |
4.7 |
| 3 |
17.95 |
20.00 |
37.10 |
97.8 |
3.4 |
4. Conclusion
In this work, a non-enzymatic H2O2 sensor based on MnO2/RGO/P25 nanocomposites has been successfully prepared using a novel photo-reduction assisted synthesis method. The as-prepared sensor exhibited highly electrocatalytic activity, selectivity and stability towards the oxidation of H2O2. Under optimum conditions, the calibration curve for H2O2 determination was linear in the range from 1.0 × 10−6 to 4.0 × 10−3 M (R2 = 0.999) with a detection limit of 3 × 10−7 M (S/N = 3). The proposed synthesis method is mild and simple, and the synthesized MnO2/RGO/P25 nanocomposites possess low toxicity. The present study provides a novel approach for the construction of non-enzyme sensors.
Acknowledgements
This research is supported by the National Natural Science Foundation of China (No. 61171033, 61571278, 61571280).
References
- S. Wolfbeis, A. Dürkop, M. Wu and Z. H. Lin, Angew. Chem., Int. Ed., 2002, 41, 4495–4498 CrossRef.
- E. C. Hurdis and H. Romeyn, Anal. Chem., 1954, 26, 320–325 CrossRef CAS.
- B. X. Li, Z. J. Zhang and Y. Jin, Sens. Actuators, B, 2001, 72, 115–119 CrossRef CAS.
- H. Cui, W. Wang, C. F. Duan, Y. P. Dong and J. Z. Guo, Chem.–Eur. J., 2007, 13, 6975–6984 CrossRef CAS PubMed.
- Y. Z. Li and A. Townshend, Anal. Chim. Acta, 1998, 359, 149–156 CrossRef CAS.
- K. Sunil and B. Narayana, Bull. Environ. Contam. Toxicol., 2008, 81, 422–426 CrossRef CAS PubMed.
- S. Y. Xu, B. Peng and X. Z. Han, Biosens. Bioelectron., 2007, 22, 1807–1810 CrossRef CAS PubMed.
- Q. Rui, K. K. Komori, Y. Tian, H. Q. Liu, Y. P. Luo and Y. Sakai, Anal. Chim. Acta, 2010, 670, 57–62 CrossRef CAS PubMed.
- S. Liu, L. M. Li, Q. Y. Hao, X. M. Yin, M. Zhang, Q. H. Li, L. B. Chen and T. H. Wang, Talanta, 2010, 81, 727–731 CrossRef CAS PubMed.
- X. B. Lu, J. H. Zhou, W. Lu, Q. Liu and J. H. Li, Biosens. Bioelectron., 2008, 23, 1236–1243 CrossRef CAS PubMed.
- C. H. Wang, C. Yang, Y. Y. Song, W. Gao and X. H. Xia, Adv. Funct. Mater., 2005, 15, 1267–1275 CrossRef CAS.
- L. Zhang, Biosens. Bioelectron., 2008, 23, 1610–1615 CrossRef CAS PubMed.
- L. M. Li, Z. F. Du, S. Liu, Q. Y. Hao, Y. G. Wang, Q. H. Li and T. H. Wang, Talanta, 2010, 82, 1637–1641 CrossRef CAS PubMed.
- H. Yao, H. Liu, M. Sun and L. Gong, Microchim. Acta, 2012, 177, 31–37 CrossRef CAS.
- J. Narang, N. Chauhan and C. S. Pundir, Analyst, 2011, 136, 4460–4466 RSC.
- S. Gupta and R. Prakash, J. Mater. Chem. C, 2014, 2, 6859–6866 RSC.
- S. Gupta and R. Prakash, RSC Adv., 2015, 5, 81660–81667 RSC.
- S. S. Kumar, J. Joseph and K. L. Phani, Chem. Mater., 2007, 19, 4722–4730 CrossRef CAS.
- W. Li, L. Kuai, Q. Qin and B. Geng, J. Mater. Chem. A, 2013, 1, 7111–7117 CAS.
- G. S. Cao, P. Wang, X. Li, Y. Wang, G. Wang and J. Li, Nano Lett., 2014, 9, 16–18 CrossRef CAS PubMed.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- K. L. Wu, X. Z. Li, C. Dong, L. Liu, P. D. Liu, T. H. Ding, J. Lu and X. W. Wei, Chem. Lett., 2013, 42, 1466–1468 CrossRef CAS.
- X. Zhang, J. Zhang, D. Zhou and G. Wang, Micro Nano Lett., 2012, 7, 60–63 CAS.
- D. X. Ye, H. X. Li, G. H. Liang, J. Luo, X. X. Zhang, S. Zhang, H. Chen and J. L. Kong, Electrochim. Acta, 2013, 109, 195–200 CrossRef CAS.
- F. Xiao, Y. Q. Li, X. L. Zan, K. Liao, R. Xu and H. W. Duan, Adv. Funct. Mater., 2012, 22, 2487–2494 CrossRef CAS.
- J. Y. Lei, X. F. Lu, W. Wang, X. J. Bian, Y. P. Xue, C. Wang and L. J. Li, RSC Adv., 2012, 2, 2541–2544 RSC.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Z. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, H. A. Margarita, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- X. Wang, L. J. Zhi and K. Müllen, Nano Lett., 2008, 8, 323–327 CrossRef CAS PubMed.
- Y. Zhou, Q. L. Bao, B. Varghese, L. Ai, L. Tang, C. K. Tan, C. H. Sow and K. P. Loh, Adv. Mater., 2010, 22, 67–71 CrossRef CAS PubMed.
- C. Liu, Y. Teng, R. Liu, S. Luo, Y. Tang, L. Chen and Q. Cai, Carbon, 2011, 49, 5312–5320 CrossRef CAS.
- B. Li, X. T. Zhang, X. H. Li, L. Wang, R. Y. Han, B. B. Liu, W. T. Zheng, X. L. Li and Y. C. Liu, Chem. Commun., 2010, 46, 3499–3501 RSC.
- S. Kalathil, M. M. Khan, S. A. Ansari, J. Lee and M. H. Cho, Nanoscale, 2013, 5, 6323–6326 RSC.
- M. M. Khan, S. A. Ansari, M. I. Amal, J. Lee and M. H. Cho, Nanoscale, 2013, 5, 4427–4435 RSC.
- H. Ishiguro, R. Nakano, Y. Y. Yao, J. Kajioka, A. Fujishima, K. Sunada, M. Minoshima, K. Hashimotod and Y. Kubota, Photochem. Photobiol. Sci., 2011, 10, 1825–1829 CAS.
- M. A. Zwijnenburg, S. T. Bromley, J. C. Jansen and T. Maschmeyer, Chem. Mater., 2004, 16, 12–20 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS PubMed.
- A. Ajmal, I. Majeed, R. N. Malik, H. Idrissc and M. A. Nadeem, RSC Adv., 2014, 4, 37003–37026 RSC.
- Y. Y. Zhu, Y. J. Wang, W. Q. Yao, R. L. Zong and Y. F. Zhu, RSC Adv., 2015, 5, 29201–29208 RSC.
- Y. L. Liu, K. Q. Chen, M. Y. Xiong, P. Zhou, Z. Y. Peng, G. J. Yang, Y. Q. Cheng, R. B. Wang and W. Chen, RSC Adv., 2014, 4, 43760–43765 RSC.
- H. K. Jeong, Y. P. Lee, R. J. Lahaye, M. H. Park, M. H. An, I. J. Kim, C. W. Yang, C. Y. Park, R. S. Ruoff and Y. H. Lee, J. Am. Chem. Soc., 2008, 130, 1362–1366 CrossRef CAS PubMed.
- C. D. Wanger, W. M. Riggs, L. E. Davis, J. F. Moulder and G. E. Muilenberg, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer Corp., Physical Electronics Division, Eden Prairie, Minnesota, USA, 1979 Search PubMed.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prudhomme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- Y. H. Zhang, Z. Tang, X. Z. Fu and Y. J. Xu, ACS Nano, 2011, 5, 7426–7435 CrossRef CAS PubMed.
- C. Julien, M. Massot, S. Rangan, M. Lemal and D. Guyomard, J. Raman Spectrosc., 2002, 33, 223–228 CrossRef.
- G. S. Shao, X. J. Zhang and Z. Y. Yuan, Appl. Catal., B, 2008, 82, 208–218 CrossRef CAS.
- H. H. Wang, Y. Bu, W. L. Dai, K. Li, H. D. Wang and X. Zuo, Sens. Actuators, B, 2015, 216, 298–306 CrossRef CAS.
- Y. H. Bai, Y. Du, J. J. Xu and H. Y. Chen, Electrochem. Commun., 2007, 9, 2611–2616 CrossRef CAS.
- B. Xu, M. L. Ye, Y. X. Yu and W. D. Zhang, Anal. Chim. Acta, 2010, 674, 20–26 CrossRef CAS PubMed.
- S. X. Deng, D. Sun, C. H. Wu, H. Wang, J. B. Liu, Y. X Sun and H. Yan, Electrochim. Acta, 2013, 111, 707–712 CrossRef CAS.
- L. M. Li, Z. F. Du, S. Liu, Q. Y. Hao, Y. G. Wang, Q. H. Li and T. H. Wang, Talanta, 2010, 82, 1637–1641 CrossRef CAS PubMed.
- B. Xu, M. L. Ye, Y. X. Yu and W. D. Zhang, Anal. Chim. Acta, 2010, 674, 20–26 CrossRef CAS PubMed.
- S. J. He, B. Y. Zhang, M. M. Liu and W. Chen, RSC Adv., 2014, 4, 49315–49323 RSC.
- D. X. Ye, H. X. Li, G. H. Liang, J. Luo, X. X. Zhang, S. Zhang, H. Chen and J. L. Kong, Electrochim. Acta, 2013, 109, 195–200 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.