
A reversible rhodamine 6G-based fluorescence turn-on probe for Fe3+ in water and its application in living cell imaging†
Abstract
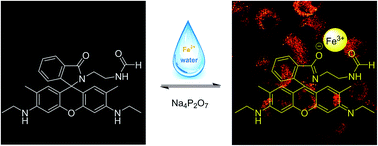
A reversible fluorescent probe L based on rhodamine 6G is synthesized for the optical detection of Fe3+ in water. This probe displays favorable selectivity for Fe3+ with a 1 : 1 stoichiometry. It displays a fluorescence turn-on response in the process of rhodamine ring opening with Fe3+ and a fluorescence turn-off response with the addition of Na4P2O7 to the L-Fe3+ complex. The association constant between the probe and Fe3+ was detected to be 3.5 × 104 M−1, and the corresponding detection limit was calculated to be 1.9 × 10−8 M according to fluorescence titration analysis. Furthermore, bioimaging investigations indicated that this probe was cell permeable and suitable for monitoring intracellular Fe3+ in living cells by confocal microscopy.
 Please wait while we load your content...
Please wait while we load your content...

